Homologous BGC Clusterization
Overview
Teaching: 40 min
Exercises: 10 minQuestions
How can I identify Gene Cluster Families?
How can I predict the production of similar metabolites
How can I clusterize BGCs into groups that produce similar metabolites?
How can I compare the metabolic capability of different bacterial lineages?
Objectives
Understand the purpose of using
BiG-SLiCEandBiG-FAMLearn the inputs required to run
BiG-SLiCEandBiG-FAMRun an example with our Streptococcus data on both softwares
Understand the obtained results
BiG-SLiCE and BiG-FAM a set of tools to compare the microbial metabolic diversity
Around the curriculum, we have been learning about the metabolic capability of bacteria lineages encoded in BGCs. Before we start this lesson, let’s remember that the products of these clusters are biomolecules that are (mostly) not essential for life. They take the role of “tools” that offer some ecological and physiological advantage to the bacterial lineage producing it.
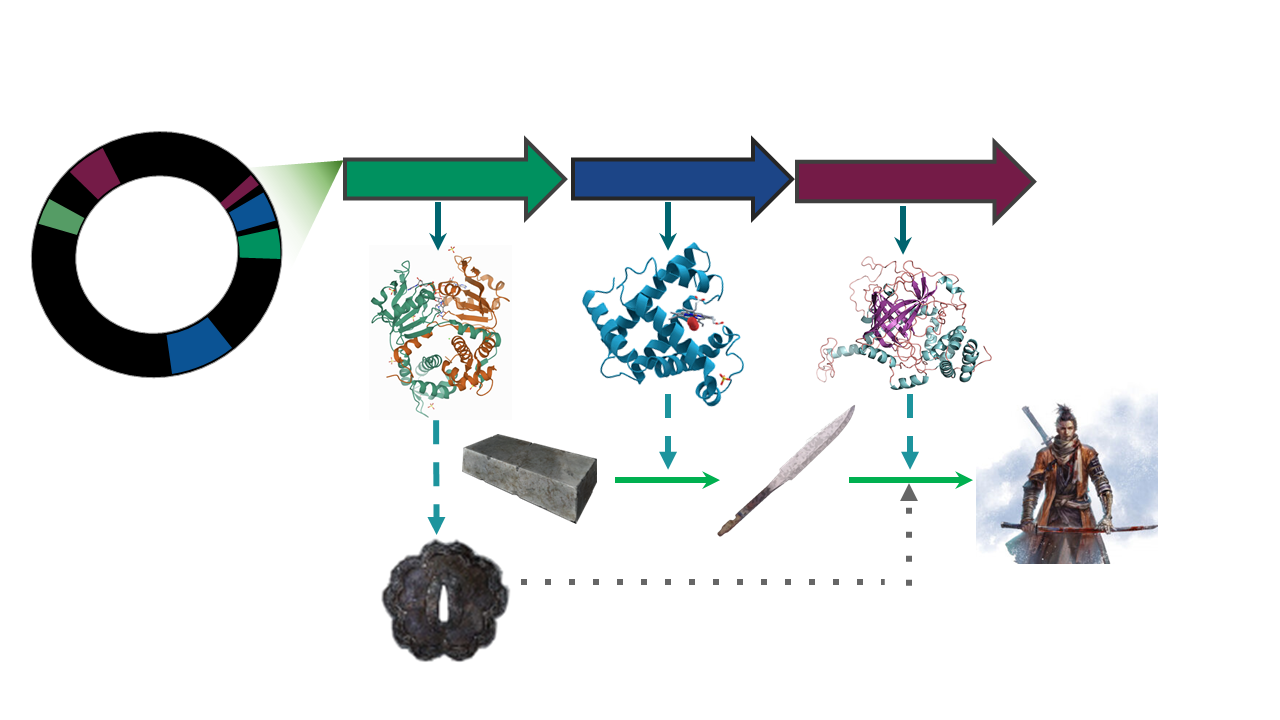
The presence/absence of a set of BGCs in a genome, can be associated to the ecological niches and (micro)environments which the lineage faces.
The counterpart of this can be taken as how biosynthetically diverse a bacterial lineage can be due to its environment.
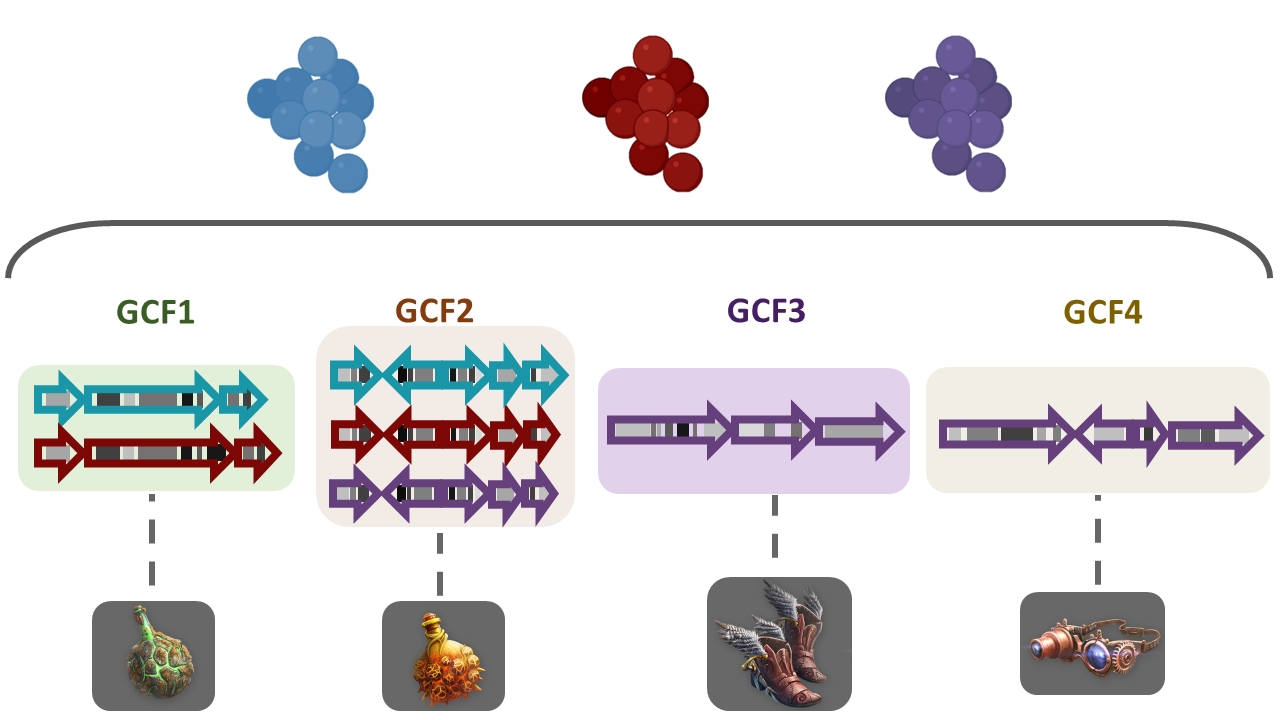
To answer some of this questions, we need a way to compare the metabolic capabilities of a set of diverse bacterial lineages. One way to do this is to compare the state (identity) of homologous BGCs. The homologous clusters that share a similar domain architecture and in consequence will produce a similar metabolite, are grouped into a bigger group that we call Gene Cluster Families (GCFs).
There are some programs capable of clustering BGCs
in GCFs, e.g. ClusterFinder and DeepBGC. BiG-SLICE is a
software that has been designed to do this work in less time, even
when dealing with large datasets (1.2 million BGCs). This
characteristic makes it a good tool to compare the metabolic
diversity of several bacterial lineages.
The input for BiG-SLiCE
Firstly, activate the conda environment, where BiG-SLiCE
has been installed as well as all the softwares required for its usage:
$ conda activate GenomeMining_Global
You will have now a (GenomeMining) label at the beginning of the
prompt line.
If we seek for help using the --help flag of bigslice, we can get
a first glance at the input that we need:
$ bigslice --help | head -n20
usage: bigslice [-i <folder_path>] [--resume] [--complete] [--threshold T]
[--threshold_pct <N>] [--query <folder_path>]
[--query_name <name>] [--run_id <id>] [--n_ranks N_RANKS]
[-t <N>] [--hmmscan_chunk_size <N>] [--subpfam_chunk_size <N>]
[--extraction_chunk_size EXTRACTION_CHUNK_SIZE] [--scratch]
[-h] [--program_db_folder PROGRAM_DB_FOLDER] [--version]
<output_folder_path>
_________
___ _____|\____|\____|\ \____ \
| \ | } } } ___)_ _/__
| > | _--/--|/----|/----|/__/ __|| __|
| < | || __/ ( (| | \___/ /__/| _|
| > || || |_ | _) ) |_ | |\ \__ | |__
|____/|_| \___/|___/|___||_| \____||____| [ Version 1.1.0 ]
Biosynthetic Gene clusters - Super Linear Clustering Engine
(https://github.com/medema-group/bigslice)
positional arguments:
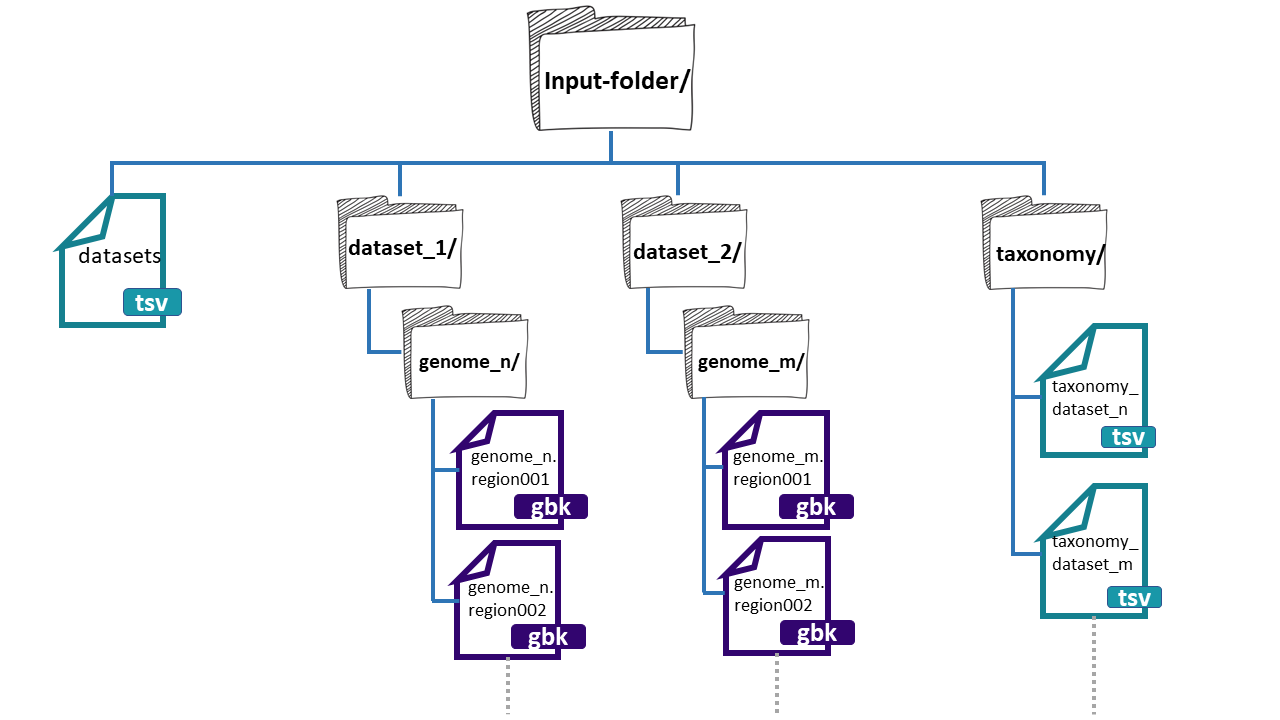
BiG-SLiCE is asking for a folder as the input. It is
important to highlight that the bones of this folder are the BGCs
from a group of genomes that has been obtained by antiSMASH. In its GitHub page, we can search for
an input folder template. The folder needs three
main components:
- datasets.tsv: This is a file that needs to have this exact name. This must be a tabular separated file where the information of each dataset (BGCs from a group of genomes) must be specified.

-
dataset_n: Each dataset will be composed by the BGCs of different genomes. We can give
BiG-SLiCEn groups of BGCs. The separation of the BGCs in different datasets is due to the different characteristics that the bacterial lineages can have. For example, if we have a set of bacterial populations that came from maize roots and other obtained from tomato roots, we can put them into dataset_1 and dataset_2 respectively. -
taxonomy_n.tsv:This is also a tabular separated file that will carry the taxonomic information of each of the genomes where the input BGCs were found and the path to reach these BGCs inside the input-folder:
- Genome folder name (ends with ‘/’)
- Kingdom / Domain name
- Class name
- Order name
- Family name
- Genus name
- Species name
- Organism / Strain name
Here, we have an example of the structure of the input-folder:
Creating the input-folder
We will create a bigslice folder inside the
results directory.
$ cd ~/gm_workshop/results/genome_mining
$ mkdir bigslice
$ cd bigslice
Now that we know what input BiG-SLiCE requires, we will do our
input-folder step by step. First, let’s remember how many genomes
we have:
$ ls -F ../antismash
agalactiae_18RS21/ agalactiae_A909/ agalactiae_COH1/
agalactiae_515/ agalactiae_CJB111/ agalactiae_H36B/
athermophilus_LMD-9/ athermophilus_LMG_18311/
We have six Streptococcus genomes from the original paper, and two
public genomes. We will allocate their respective BGC .gbks in
two datasets, one for the genomes from Tettelin et al. paper, and
the other for the public ones.
$ mkdir input-folder
$ mkdir input-folder/dataset_1
$ mkdir input-folder/dataset_2
$ mkdir input-folder/taxonomy
$ tree
.
└── input-folder
├── dataset_1
├── dataset_2
└── taxonomy
4 directories, 0 files
Now we will create the folders for the BGCs of the dataset_1 genomes. We will take advantage of the strain name of our Streptococcus agalactiae lineages to create their folders:
$ for i in 18RS21 COH1 515 H36B A909 CJB111;
do mkdir input-folder/dataset_1/Streptococcus_agalactiae_$i;
done
$ tree -F
.
└── input-folder/
├── dataset_1/
│ ├── Streptococcus_agalactiae_18RS21/
│ ├── Streptococcus_agalactiae_515/
│ ├── Streptococcus_agalactiae_A909/
│ ├── Streptococcus_agalactiae_CJB111/
│ ├── Streptococcus_agalactiae_COH1/
│ └── Streptococcus_agalactiae_H36B/
├── dataset_2/
└── taxonomy/
10 directories, 0 files
We will do the same for the public genomes:
$ for i in LMD-9 LMG_18311;
do mkdir input-folder/dataset_2/Streptococcus_thermophilus_$i;
done
$ tree -F
.
└── input-folder/
├── dataset_1/
│ ├── Streptococcus_agalactiae_18RS21/
│ ├── Streptococcus_agalactiae_515/
│ ├── Streptococcus_agalactiae_A909/
│ ├── Streptococcus_agalactiae_CJB111/
│ ├── Streptococcus_agalactiae_COH1/
│ └── Streptococcus_agalactiae_H36B/
├── dataset_2/
│ ├── Streptococcus_thermophilus_LMD-9/
│ └── Streptococcus_thermophilus_LMG_18311/
└── taxonomy/
12 directories, 0 files
Next, we will copy the BGCs .gbk files from the dataset_1
genomes from each AntiSMASH output directory to its respective
folder .
$ ls input-folder/dataset_1/ | while read line;
do cp ../antismash/output/$line/*region*.gbk input-folder/dataset_1/$line;
done
$ tree -F
.
└── input-folder/
├── dataset_1/
│ ├── Streptococcus_agalactiae_18RS21/
│ │ ├── AAJO01000016.1.region001.gbk
│ │ ├── AAJO01000043.1.region001.gbk
│ │ └── AAJO01000226.1.region001.gbk
│ ├── Streptococcus_agalactiae_515/
│ │ ├── AAJP01000027.1.region001.gbk
│ │ └── AAJP01000037.1.region001.gbk
│ ├── Streptococcus_agalactiae_A909/
│ │ ├── CP000114.1.region001.gbk
│ │ └── CP000114.1.region002.gbk
│ ├── Streptococcus_agalactiae_CJB111/
│ │ ├── AAJQ01000010.1.region001.gbk
│ │ └── AAJQ01000025.1.region001.gbk
│ ├── Streptococcus_agalactiae_COH1/
│ │ ├── AAJR01000002.1.region001.gbk
│ │ └── AAJR01000044.1.region001.gbk
│ └── Streptococcus_agalactiae_H36B/
│ ├── AAJS01000020.1.region001.gbk
│ └── AAJS01000117.1.region001.gbk
├── dataset_2/
│ ├── Streptococcus_thermophilus_LMD-9/
│ └── Streptococcus_thermophilus_LMG_18311/
└── taxonomy/
12 directories, 13 files
Again, we will do the same for the BGCs .gbks of the dataset_2:
$ ls input-folder/dataset_2/ | while read line;
do cp ../antismash/output/$line/*region*.gbk input-folder/dataset_2/$line;
done
$ tree -F
.
└── input-folder/
├── dataset_1/
│ ├── Streptococcus_agalactiae_18RS21/
│ │ ├── AAJO01000016.1.region001.gbk
│ │ ├── AAJO01000043.1.region001.gbk
│ │ └── AAJO01000226.1.region001.gbk
│ ├── Streptococcus_agalactiae_515/
│ │ ├── AAJP01000027.1.region001.gbk
│ │ └── AAJP01000037.1.region001.gbk
│ ├── Streptococcus_agalactiae_A909/
│ │ ├── CP000114.1.region001.gbk
│ │ └── CP000114.1.region002.gbk
│ ├── Streptococcus_agalactiae_CJB111/
│ │ ├── AAJQ01000010.1.region001.gbk
│ │ └── AAJQ01000025.1.region001.gbk
│ ├── Streptococcus_agalactiae_COH1/
│ │ ├── AAJR01000002.1.region001.gbk
│ │ └── AAJR01000044.1.region001.gbk
│ └── Streptococcus_agalactiae_H36B/
│ ├── AAJS01000020.1.region001.gbk
│ └── AAJS01000117.1.region001.gbk
├── dataset_2/
│ ├── Streptococcus_thermophilus_LMD-9/
│ │ ├── CP000419.1.region001.gbk
│ │ ├── CP000419.1.region002.gbk
│ │ ├── CP000419.1.region003.gbk
│ │ ├── CP000419.1.region004.gbk
│ │ └── CP000419.1.region005.gbk
│ └── Streptococcus_thermophilus_LMG_18311/
│ ├── NC_006448.1.region001.gbk
│ ├── NC_006448.1.region002.gbk
│ ├── NC_006448.1.region003.gbk
│ ├── NC_006448.1.region004.gbk
│ ├── NC_006448.1.region005.gbk
│ └── NC_006448.1.region006.gbk
└── taxonomy/
12 directories, 24 files
Exercise 1. Input-folder structure
As we have seen, the structure of the input folder for BiG-SLiCE is quite difficult to get. Imagine “Sekiro” wants to copy the directory structure to use it for future BiG-SLiCE inputs. Consider the following directory structure:
└── input_folder/ ├── datasets.tsv ├── dataset_1/ | ├── genome_1A/ | | ├── genome_1A.region001.gbk | | └── genome_1A.region002.gbk | ├── genome_1B/ | | ├── genome_1B.region001.gbk | | └── genome_1B.region002.gbk ├── dataset_2/ | ├── genome_2A/ | | ├── genome_2A.region001.gbk | | └── genome_2A.region002.gbk | ├── genome_2B/ | | ├── genome_2B.region001.gbk | | └── genome_2B.region002.gbk └── taxonomy/ ├── taxonomy_dataset_1.tsv └── taxonomy_dataset_2.tsvWhich would be the commands to copy the directory structure without the files (just the folders)?
a)$ cp -r input_folder input_folder_copy $ cd input_folder_copy $ rm -rb)
$ mv -r input_folder input_folder_copy $ rm input_folder_copy/dataset_1/genome_1A/* input_folder_copy/dataset_1/genome_1B/* $ rm input_folder_copy/dataset_2/genome_2A/* input_folder_copy/dataset_2/genome_2B/* $ rm input_folder_copy/taxonomy/*c)
$ cp -r input_folder input_folder_copy $ rm input_folder_copy/dataset_1/genome_1A/* input_folder_copy/dataset_1/genome_1B/* $ rm input_folder_copy/dataset_2/genome_2A/* input_folder_copy/dataset_2/genome_2B/* $ rm input_folder_copy/taxonomy/*d)
$ cp -r input_folder input_folder_copy $ rm input_folder_copy/dataset_1/genome_1A/* /genome_1B/* $ rm input_folder_copy/dataset_2/genome_2A/* /genome_2B/* $ rm input_folder_copy/taxonomy/*Solution
First we need to copy the whole directory to make an input_folder_copy. After that we want to erase the files inside the directories dataset_1/genome_1A, dataset_1/genome_1B, dataset_2/genome_2A, dataset2/genome_2B and taxonomy/
c)$ cp -r input_folder input_folder_copy $ rm input_folder_copy/dataset_1/genome_1A/* input_folder_copy/dataset_1/genome_1B/* $ rm input_folder_copy/dataset_2/genome_2A/* input_folder_copy/dataset_2/genome_2B/* $ rm input_folder_copy/taxonomy/*
datasets.tsv file - The silmaril of BiG-SLiCE
The next step is to create the main file datasets.tsv. First we
put the first lines to this file. In each of these .tsv files, we
can put lines that begin with #. This will not be read by the
code, and it can be of help to know which information is located
in those files. We will use the command echo with the -e flag
that will enable us to put tab separations between the text:
$ cd input-folder
$ echo -e "#Dataset name""\t""Path to folder""\t""Path to taxonomy""\t""Description" > datasets.tsv
$ more datasets.tsv
#Dataset name Path to folder Path to taxonomy Description
We will use echo One more time to put the information of our two
datasets. But this time, we will indicate bash that we want to insert
this new line in the existing datasets.tsv with the >> option.
$ echo -e "dataset_1""\t""dataset_1/""\t""taxonomy/dataset_1_taxonomy.tsv""\t""Tetteling genomes" >> datasets.tsv
$ echo -e "dataset_2""\t""dataset_2/""\t""taxonomy/dataset_2_taxonomy.tsv""\t""Public genomes" >> datasets.tsv
$ more datasets.tsv
#Dataset name Path to folder Path to taxonomy Description
dataset_1 dataset_1/ taxonomy/dataset_1_taxonomy.tsv Tetteling genomes
dataset_2 dataset_2/ taxonomy/dataset_2_taxonomy.tsv Public genomes
The main file of the input-folder is finished!
The taxonomy of the input-folder
We also need to specify the taxonomic assignation of each of the
input genomes BGC’s. We will build one dataset_taxonomy.tsv file
for each one of our datasets, i.e. two in total. Firstly, create
the header of each one of these files using echo:
$ echo -e "#Genome folder""\t"Kingdom"\t"Phylum"\t"Class"\t"Order"\t"Family"\t"Genus"\t""Species Organism" > taxonomy/dataset_1_taxonomy.tsv
$ echo -e "#Genome folder""\t"Kingdom"\t"Phylum"\t"Class"\t"Order"\t"Family"\t"Genus"\t""Species Organism" > taxonomy/dataset_2_taxonomy.tsv
$ more taxonomy/dataset_1_taxonomy.tsv
#Genome folder Kingdom Phylum Class Order Family Genus Species Organism
There are several ways to obtain the taxonomic data. One of them is
searching for each lineage taxonomic identification on NCBI. There is also a way to get this information from our .gbk files:
$ head -n20 ../../annotated/Streptococcus_agalactiae_18RS21.gbk
LOCUS AAJO01000169.1 2501 bp DNA linear UNK
DEFINITION Streptococcus agalactiae 18RS21
ACCESSION AAJO01000169.1
KEYWORDS .
SOURCE Streptococcus agalactiae 18RS21.
ORGANISM Streptococcus agalactiae 18RS21
Bacteria; Terrabacteria group; Firmicutes; Bacilli;
Lactobacillales; Streptococcaceae; Streptococcus; Streptococcus
agalactiae.
FEATURES Location/Qualifiers
source 1..2501
/mol_type="genomic DNA"
/db_xref="taxon:342613"
/genome_md5=""
/project="nselem35_342613"
/genome_id="342613.37"
/organism="Streptococcus agalactiae 18RS21"
CDS 1..213
/db_xref="SEED:fig|342613.37.peg.1973"
/db_xref="GO:0003735"
We will use again echo to write this information into our
dataset_taxonomy.tsv. This is going to be a little long because
we have 8 genomes and we will need to write 8 echo lines, but
bear with us. We are almost there!
$ echo -e "Streptococcus_agalactiae_18RS21/""\t"Bacteria"\t"Firmicutes"\t"Bacilli"\t"Lactobacillales"\t"Streptococcaceae"\t"Streptococcus"\t""Streptococcus agalactiae 18RS21" >> taxonomy/dataset_1_taxonomy.tsv
$ echo -e "Streptococcus_agalactiae_515/""\t"Bacteria"\t"Firmicutes"\t"Bacilli"\t"Lactobacillales"\t"Streptococcaceae"\t"Streptococcus"\t""Streptococcus agalactiae 515" >> taxonomy/dataset_1_taxonomy.tsv
$ echo -e "Streptococcus_agalactiae_A909/""\t"Bacteria"\t"Firmicutes"\t"Bacilli"\t"Lactobacillales"\t"Streptococcaceae"\t"Streptococcus"\t""Streptococcus agalactiae A909" >> taxonomy/dataset_1_taxonomy.tsv
$ echo -e "Streptococcus_agalactiae_CJB111/""\t"Bacteria"\t"Firmicutes"\t"Bacilli"\t"Lactobacillales"\t"Streptococcaceae"\t"Streptococcus"\t""Streptococcus agalactiae CJB111" >> taxonomy/dataset_1_taxonomy.tsv
$ echo -e "Streptococcus_agalactiae_COH1/""\t"Bacteria"\t"Firmicutes"\t"Bacilli"\t"Lactobacillales"\t"Streptococcaceae"\t"Streptococcus"\t""Streptococcus agalactiae COH1" >> taxonomy/dataset_1_taxonomy.tsv
$ echo -e "Streptococcus_agalactiae_H36B/""\t"Bacteria"\t"Firmicutes"\t"Bacilli"\t"Lactobacillales"\t"Streptococcaceae"\t"Streptococcus"\t""Streptococcus agalactiae H36B" >> taxonomy/dataset_1_taxonomy.tsv
$ more taxonomy/dataset_1_taxonomy.tsv
#Genome folder Kingdom Phylum Class Order Family Genus Species Organism
Streptococcus_agalactiae_18RS21/ Bacteria Firmicutes Bacilli Lactobacillales Strept
ococcaceae Streptococcus Streptococcus agalactiae 18RS21
Streptococcus_agalactiae_515/ Bacteria Firmicutes Bacilli Lactobacillales Streptococcace
ae Streptococcus Streptococcus agalactiae 515
Streptococcus_agalactiae_A909/ Bacteria Firmicutes Bacilli Lactobacillales Streptococcace
ae Streptococcus Streptococcus agalactiae A909
Streptococcus_agalactiae_CJB111/ Bacteria Firmicutes Bacilli Lactobacillales Strept
ococcaceae Streptococcus Streptococcus agalactiae CJB111
Streptococcus_agalactiae_COH1/ Bacteria Firmicutes Bacilli Lactobacillales Streptococcace
ae Streptococcus Streptococcus agalactiae COH1
Streptococcus_agalactiae_H36B/ Bacteria Firmicutes Bacilli Lactobacillales Streptococcace
ae Streptococcus Streptococcus agalactiae H36B
We will do the same for the dataset_2_taxonomy.tsv file:
$ echo -e "Streptococcus_thermophilus_LMD-9/""\t"Bacteria"\t"Firmicutes"\t"Bacilli"\t"Lactobacillales"\t"Streptococcaceae"\t"Streptococcus"\t""Streptococcus thermophilus LMD-9" >> taxonomy/dataset_2_taxonomy.tsv
$ echo -e "Streptococcus_thermophilus_LMG_18311/""\t"Bacteria"\t"Firmicutes"\t"Bacilli"\t"Lactobacillales"\t"Streptococcaceae"\t"Streptococcus"\t""Streptococcus thermophilus LMG 18311" >> taxonomy/dataset_2_taxonomy.tsv
$ more taxonomy/dataset_2_taxonomy.tsv
#Genome folder Kingdom Phylum Class Order Family Genus Species Organism
Streptococcus_thermophilus_LMD-9/ Bacteria Firmicutes Bacilli Lactobacillales Strept
ococcaceae Streptococcus Streptococcus thermophilus LMD-9
Streptococcus_thermophilus_LMG_18311/ Bacteria Firmicutes Bacilli Lactobacillales Strept
ococcaceae Streptococcus Streptococcus thermophilus LMG 18311
Now, we have the organization of our input-folder complete
$ cd ..
$ tree -F input-folder/
input-folder/
├── dataset_1/
│ ├── Streptococcus_agalactiae_18RS21/
│ │ ├── AAJO01000016.1.region001.gbk
│ │ ├── AAJO01000043.1.region001.gbk
│ │ └── AAJO01000226.1.region001.gbk
│ ├── Streptococcus_agalactiae_515/
│ │ ├── AAJP01000027.1.region001.gbk
│ │ └── AAJP01000037.1.region001.gbk
│ ├── Streptococcus_agalactiae_A909/
│ │ ├── CP000114.1.region001.gbk
│ │ └── CP000114.1.region002.gbk
│ ├── Streptococcus_agalactiae_CJB111/
│ │ ├── AAJQ01000010.1.region001.gbk
│ │ └── AAJQ01000025.1.region001.gbk
│ ├── Streptococcus_agalactiae_COH1/
│ │ ├── AAJR01000002.1.region001.gbk
│ │ └── AAJR01000044.1.region001.gbk
│ └── Streptococcus_agalactiae_H36B/
│ ├── AAJS01000020.1.region001.gbk
│ └── AAJS01000117.1.region001.gbk
├── dataset_2/
│ ├── Streptococcus_thermophilus_LMD-9/
│ │ ├── CP000419.1.region001.gbk
│ │ ├── CP000419.1.region002.gbk
│ │ ├── CP000419.1.region003.gbk
│ │ ├── CP000419.1.region004.gbk
│ │ └── CP000419.1.region005.gbk
│ └── Streptococcus_thermophilus_LMG_18311/
│ ├── NC_006448.1.region001.gbk
│ ├── NC_006448.1.region002.gbk
│ ├── NC_006448.1.region003.gbk
│ ├── NC_006448.1.region004.gbk
│ ├── NC_006448.1.region005.gbk
│ └── NC_006448.1.region006.gbk
├── datasets.tsv
└── taxonomy/
├── dataset_1_taxonomy.tsv
└── dataset_2_taxonomy.tsv
11 directories, 27 files
Exercise 2. Taxonomic information
If you use the head -n20 on one of your .gbk files, you will obtain an output like this
LOCUS AAJO01000169.1 2501 bp DNA linear UNK DEFINITION Streptococcus agalactiae 18RS21 ACCESSION AAJO01000169.1 KEYWORDS . SOURCE Streptococcus agalactiae 18RS21. ORGANISM Streptococcus agalactiae 18RS21 Bacteria; Terrabacteria group; Firmicutes; Bacilli; Lactobacillales; Streptococcaceae; Streptococcus; Streptococcus agalactiae. FEATURES Location/Qualifiers source 1..2501 /mol_type="genomic DNA" /db_xref="taxon:342613" /genome_md5="" /project="nselem35_342613" /genome_id="342613.37" /organism="Streptococcus agalactiae 18RS21" CDS 1..213 /db_xref="SEED:fig|342613.37.peg.1973" /db_xref="GO:0003735"As you can see, in the gbk file we can find the taxonomic information that we need to fill the taxonomic information of each genome. Fill in the blank spaces to complete the command that would get the organism information from the .gbk file.
$ grep ___________ -m1 ____________ Streptococcus_agalactiae_18RS21.gbk
-B 3 -A 3 -a 'Streptococcus' 'ORGANISM' 'gbk'
To obtain the next result:
ORGANISM Streptococcus agalactiae 18RS21 Bacteria; Terrabacteria group; Firmicutes; Bacilli; Lactobacillales; Streptococcaceae; Streptococcus; Streptococcus agalactiae.You can use the
grep --helpcommand to get information about the available options for thegrepcommandSolution
We need to use the grep command to search for the word “ORGANISM” in the file. Once located we just want to get the first instance of the word that is why we use the -m1 flag. Then we want the 3 next lines of the file after the instance, because that’s where the information is. So the final command would be:
$ grep 'ORGANISM' -m1 -A3 Streptococcus_agalactiae_18RS21.gbk
Running BiG-SLiCE
We are ready to run BiG-SLICE. After we type the next line
of command, it will take close to 3 minutes long to end the
process:
$ bigslice -i input-folder output-bigslice
...
...
BiG-SLiCE run complete!
In order to see the results, we need to download the
output-bigslice to our local computer. We will use
the scp command to accomplish this.
Open another terminal and without connecting it to
the remote computer (i.e. in your own computer), move to
a directory where you know you can save the folder. In this
example we will copy it to the Documents folder of our
computer.
$ cd Documents/
$ scp -r virtual-computerbetterlab@###.###.###.##/home/virtual-User/dc_workshop/results/bigslice/output-bigslice .
$ ls -F
output-bigslice/
The BiG-SLiCE prepared the code so that in each output
folder we will obtain the output files and some scripts that
will generate a way to visualize the data.
$ ls output-bigslice/
app LICENSE.txt requirements.txt result start_server.sh
The file requirements.txt contains the names of two
softwares that need to be installed to use the visualization:
SQLite3 and flask. We will install these with the next
command:
$ pip install -r output-bigslice/requirements.txt
...
...
Successfully installed pysqlite3
After the installation, we can use the start_server.sh to
generate a
$ bash output-bigslice/start_server.sh
* Serving Flask app "/mnt/d/documentos/sideprojects/sc-mineria/bigSlice/output-bigslice/app/run.py"
* Environment: production
WARNING: This is a development server. Do not use it in a production deployment.
Use a production WSGI server instead.
* Debug mode: off
* Running on http://0.0.0.0:5000/ (Press CTRL+C to quit)
The result looks a little intimidating, but if you obtained a result that looks like the above one do not worry. Next, we need to open an internet browser. In a new tab type the next line.
http://localhost:5000/
As a result, we will obtain a web-page that looks like this:
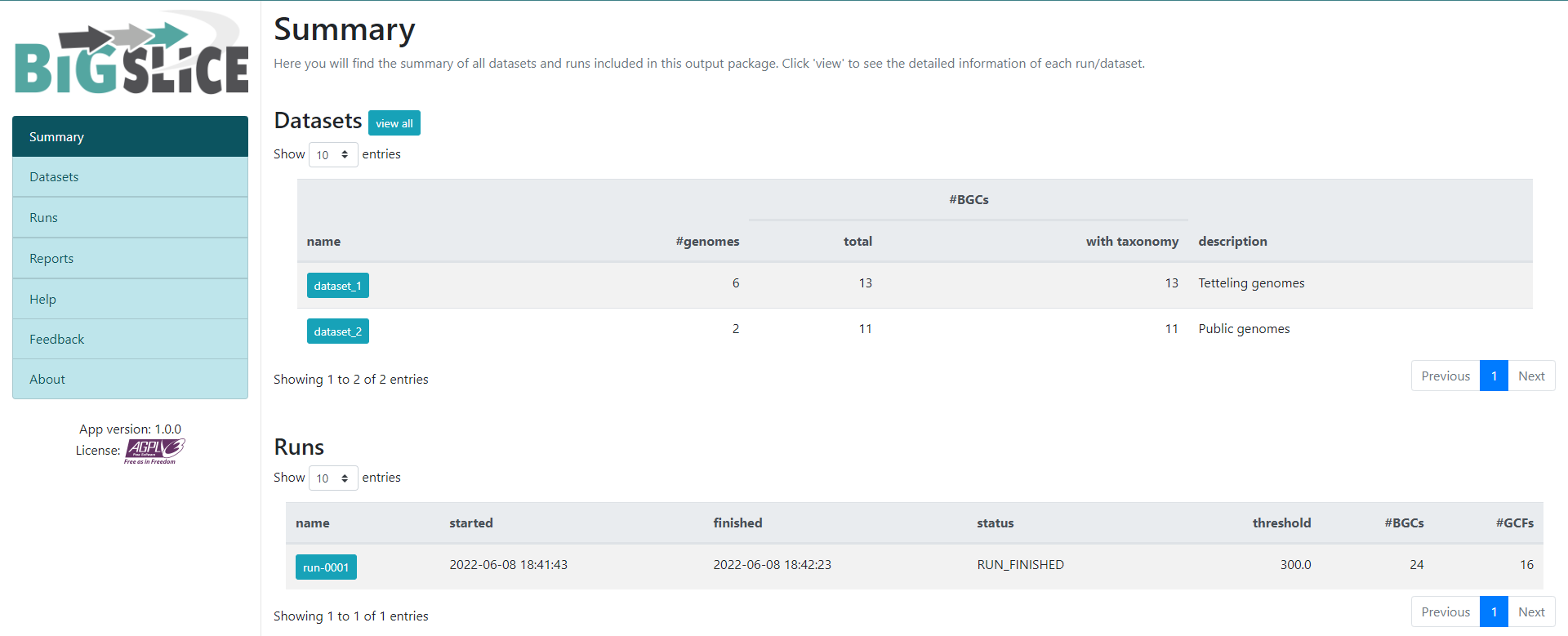
Here we have a summary of the obtained results. In the left part, we see a
set of 7 fields. 3 of them have the information that we generated: Summary,
Datasets, and Runs. The other ones are for information regarding the
software, one of them is to give feedback to the developers. You can acces to the
Datasets and Runs page also from the options that we see in the main part of
this summary page. The first part of this summary describes the datasets that
we provided as input. The second one is the information of the process (when we
ran the program) that BiG-SLiCE carry out with our data.
If we click into the Runs field on the left or the run-0001 blue button on the
Runs main section, we will jump into the page that gives us the information
regarding the BGCs GCFs that were obtained:
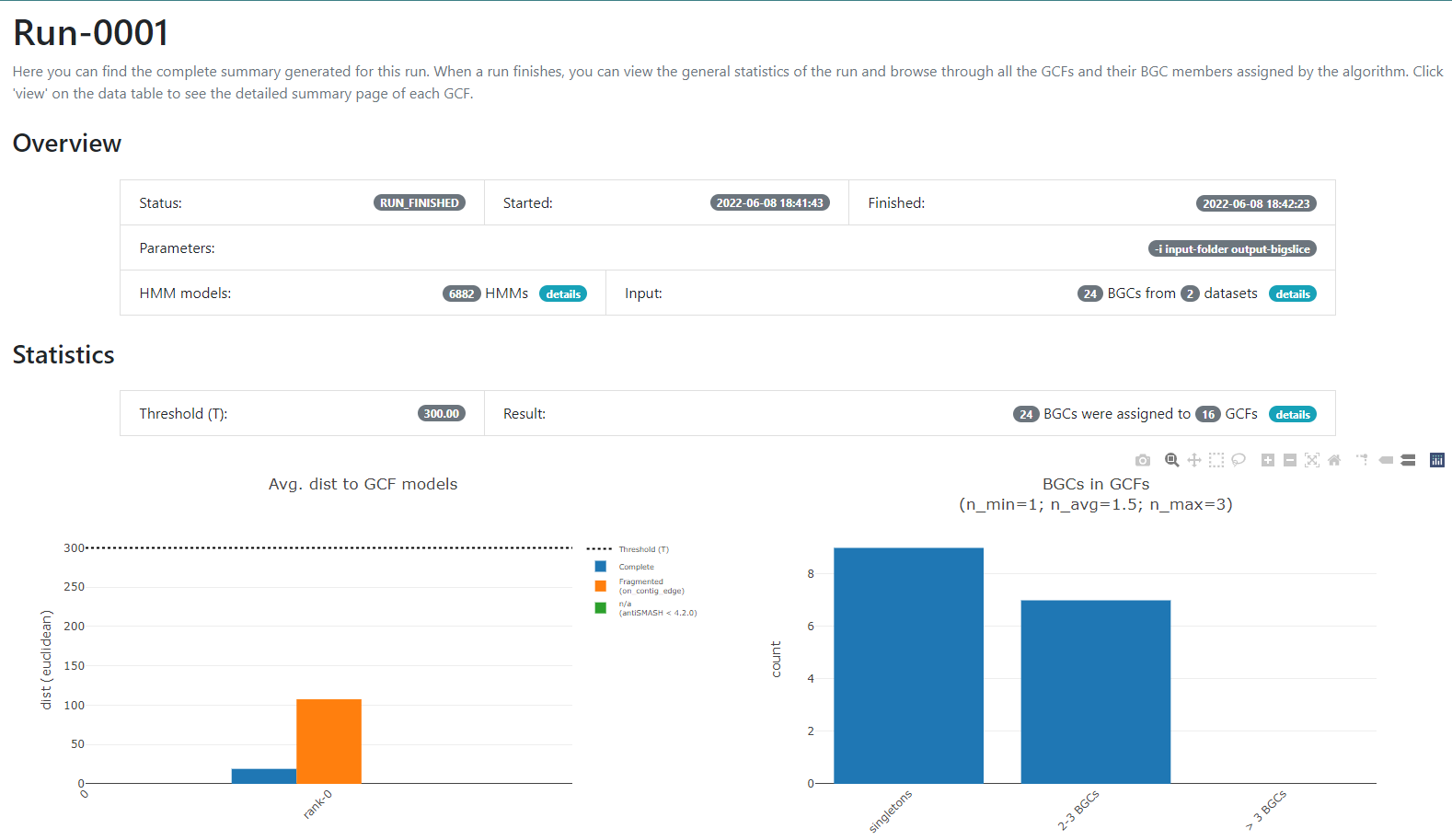
In this new page we have two bar-plots that give us information regarding if the BGCs that
we sumministrated were classified as fragmented or complete by AntiSMASH
(according to the AntiSMASH algorithm, a BGC can be classified as fragmented
if it is found at the edge of a contig), and how many families were obtained
with singletons (composed of just one BGC) and of 2-3 BGCs.
If we go to the bottom of this page, we will found a table of all the GCFs with information of the number of BGCs that compose them, the representative BGC class inside each family, and which genus has more BGCs that belong to each family.
Click the + symbol of the GCF_7, and then on the View button that
is displayed. This will take us to a page with statistics of the BGCs that
are part of this family.
Inside this section, we can found a great amount of information regarding the GCF_7
BGC’s. If we jump to section called Members, we can click on the
view BGC arrowers button to get a visualization of the domains that are
part of each of the genes of these BGCs.
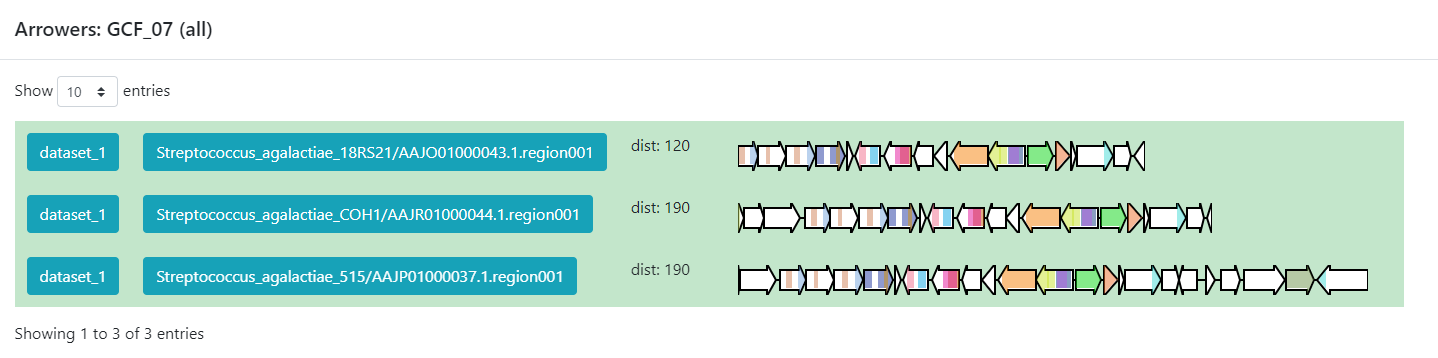
As we can see, this tool can be used to clusterize homologous BGCs that share a structure. With this knowledge, we can try some hypothesis or even build new ones in the light of this information.
Discussion
Considering the results in the runs tab, we can see that there are a lot of Gene Cluster Families that have only one cluster associated to it (GCF_1, GCF_2, GCF_4, GCF_5, GCF_6, GCF_8, GCF_9, GCF_13, and GCF_15).
What does this tell us about the metabolites diversity of the Streptococcus sample?Solution
We can see that there are some unique gene cluster families, which means that there are an organism that has that unique gene cluster and therefore it has some metabolite that the others don´t and that may be advantageous in its environment. Since there are many singleton Gene Cluster Families, we can say that there is a big diversity from the sample because many of the organisms in the sample have unique “abilities” (metabolites).
Discussion
Suppose we get the following results from a sample of 6 organisms.
GCF #core members GCF_1 6 GCF_2 5 GCF_3 5 GCF_4 6 What can we infer about the metabolite diversity?
Solution
As most of the organisms have a gene cluster in the gene cluster family, we can infer that most of them have the same secondary metabolites, which means that there is little metabolite diversity between them.
BiG-FAM
To demonstrate the functionality of BiG-SLICE, the authors gathered close to
1.2 million of BGCs. All this information, they put it on a great database that they
called BiG-FAM. When you get to the main page,
you will face a structure that looks like the one we saw on the BiG-SLICE results:
One of the options on the left is Query. If we click here, we will move to a new
page where we can insert an antiSMASH job ID. This will run a comparison of all
the BGCs found on that antiSMASH run on a single genome, against the immense database.
We will use the antiSMASH job ID from the analysis made on
Streptococcus agalactiae A909. The antiSMASH job ID can be found
in the URL direction of this page in the next format:
taxon-aaaaaaa-bbbb-cccc-dddd-eeeeeeeeeeee.
We will use the job ID that we obtained:
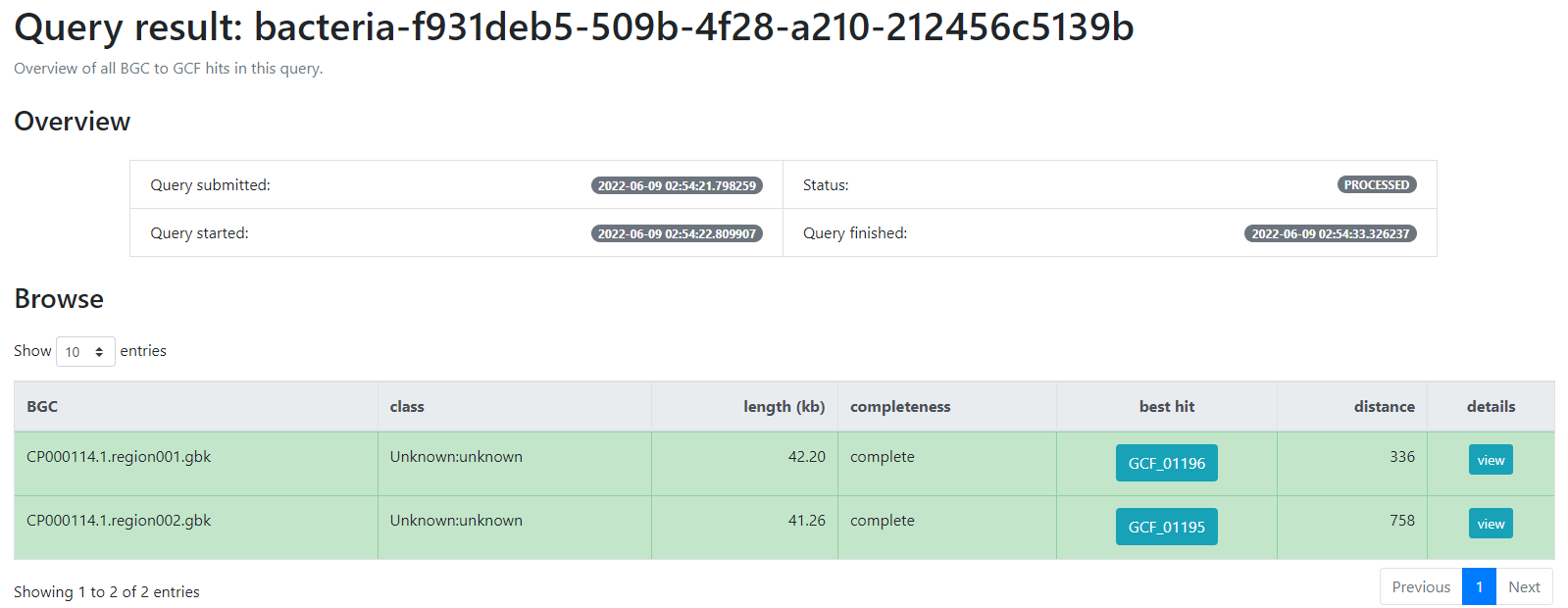
bacteria-f931deb5-509b-4f28-a210-212456c5139b
And click the submit option.
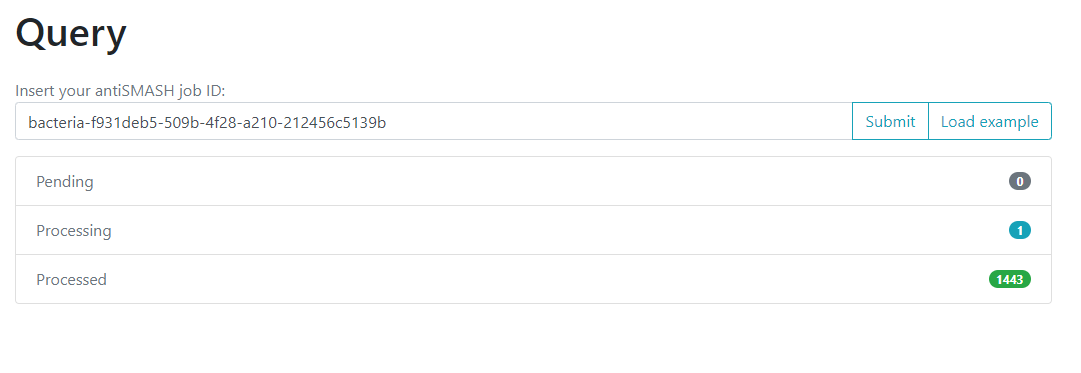
This will generate a result page that indicates with which BGCs from the database, the BGCs from the query genome are related.
Key Points
BiG-SLiCEandBiG-FAMare softwares that are useful to compare the metabolic diversity of bacterial lineages between each other and against a big databaseAn input-folder containing the BGCs from antiSMASH and the taxonomic information of each genome is needed to run
BiG-SLiCEThe results from the antiSMASH web-tool are needed to run
BiG-FAMGene Cluster Families can help us to compare the metabolic capabilities of a set of bacterial lineages
We can use
BiG-FAMto compare a BGC against the whole database and predict its Gene Cluster Family