Processes
Last updated on 2024-08-23 | Edit this page
Overview
Questions
- How do I run tasks/processes in Nextflow?
- How do I get data, files and values, into a processes?
Objectives
- Understand how Nextflow uses processes to execute tasks.
- Create a Nextflow process.
- Define inputs to a process.
Processes
We now know how to create and use Channels to send data around a workflow. We will now see how to run tasks within a workflow using processes.
A process is the way Nextflow executes commands you
would run on the command line or custom scripts.
A process can be thought of as a particular step in a workflow, e.g. an alignment step in RNA-seq analysis. Processes are independent of each other (don’t require any another process to execute) and can not communicate/write to each other. Data is passed between processes via input and output Channels.
For example, below is the command you would run to count the number of sequence records in a FASTA format file such as the yeast transcriptome:
FASTA format
FASTA format is a text-based format for representing either nucleotide sequences or peptide sequences. A sequence in FASTA format begins with a single-line description, followed by lines of sequence data. The description line is distinguished from the sequence data by a greater-than (“>”) symbol in the first column.
BASH
>YBR024W_mRNA cdna chromosome:R64-1-1:II:289445:290350:1 gene:YBR024W gene_biotype:protein_coding transcript_biotype:protein_coding gene_symbol:SCO2 description:Protein anchored to mitochondrial inner membrane; may have a redundant function with Sco1p in delivery of copper to cytochrome c oxidase; interacts with Cox2p; SCO2 has a paralog, SCO1, that arose from the whole genome duplication [Source:SGD;Acc:S000000228]
ATGTTGAATAGTTCAAGAAAATATGCTTGTCGTTCCCTATTCAGACAAGCGAACGTCTCA
ATAAAAGGACTCTTTTATAATGGAGGCGCATATCGAAGAGGGTTTTCAACGGGATGTTGTzgrep -c ‘^>’
The command
zgrep -c '^>' data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz
is used in Unix-like systems for a specific purpose: it counts the
number of sequences in a compressed FASTA file. The tool
zgrep combines the functionalities of ‘grep’ for pattern
searching and ‘gzip’ for handling compressed files. The -c
option modifies this command to count the occurrences of lines matching
the pattern, instead of displaying them. The pattern
'^>' is designed to find lines that start with ‘>’,
which in FASTA files, denotes the beginning of a new sequence. Thus,
this command efficiently counts how many sequences are contained within
the specified compressed FASTA file.
OUTPUT
6612Now we will show how to convert this into a simple Nextflow process.
Process definition
The process definition starts with keyword process,
followed by process name, in this case NUMSEQ, and finally
the process body delimited by curly brackets
{}. The process body must contain a string which represents
the command or, more generally, a script that is executed by it.
GROOVY
process NUMSEQ {
script:
"zgrep -c '^>' ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
}This process would run once.
Implicit variables
We use the Nextflow implicit variable ${projectDir} to
specify the directory where the main script is located. This is
important as Nextflow scripts are executed in a separate working
directory. A full list of implicit variables can be found here
To add the process to a workflow add a workflow block,
and call the process like a function. We will learn more about the
workflow block in the workflow episode.
GROOVY
//process_01.nf
process NUMSEQ {
script:
"zgrep -c '^>' ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
}
workflow {
//process is called like a function in the workflow block
NUMSEQ()
}We can now run the process:
Note We need to add the Nextflow run option
-process.debug to print the output to the terminal.
OUTPUT
N E X T F L O W ~ version 21.10.6
Launching `process_01.nf` [modest_pike] - revision: 3eaa812b17
executor > local (1)
[cd/eab1fd] process > NUMSEQ [100%] 1 of 1 ✔
6612GROOVY
process COUNT_BASES {
script:
"""
zgrep -v '^>' ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz|tr -d '\n'|wc -m
"""
}
workflow {
COUNT_BASES()
}OUTPUT
N E X T F L O W ~ version 21.04.0
Launching `simple_process.nf`` [prickly_gilbert] - revision: 471a79c65c
executor > local (1)
[56/5e6001] process > COUNT_BASES [100%] 1 of 1 ✔
8772368Definition blocks
The previous example was a simple process with no
defined inputs and outputs that ran only once. To control inputs,
outputs and how a command is executed a process may contain five
definition blocks:
- directives - 0, 1, or more: allow the definition of optional settings that affect the execution of the current process e.g. the number of cpus a task uses and the amount of memory allocated.
- inputs - 0, 1, or more: Define the input dependencies, usually channels, which determines the number of times a process is executed.
- outputs - 0, 1, or more: Defines the output channels used by the process to send results/data produced by the process.
- when clause - optional: Allows you to define a condition that must be verified in order to execute the process.
- script block - required: A statement within quotes that defines the commands that are executed by the process to carry out its task.
The syntax is defined as follows:
Script
At minimum a process block must contain a script
block.
The script block is a String “statement” that defines
the command that is executed by the process to carry out its task. These
are normally the commands you would run on a terminal.
A process contains only one script block, and it must be
the last statement when the process contains input and
output declarations.
The script block can be a simple one line string in
quotes e.g.
GROOVY
process NUMSEQ {
script:
"zgrep -c '^>' ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
}
workflow {
NUMSEQ()
}Or, for commands that span multiple lines you can encase the command
in triple quotes """.
For example:
GROOVY
//process_multi_line.nf
process NUMSEQ_CHR {
script:
"""
zgrep '^>' ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz > ids.txt
zgrep -c '>YA' ids.txt
"""
}
workflow {
NUMSEQ_CHR()
}OUTPUT
N E X T F L O W ~ version 21.10.6
Launching `process_multi_line.nf` [focused_jang] - revision: e32caf0dcb
executor > local (1)
[90/6e38f4] process > NUMSEQ_CHR [100%] 1 of 1 ✔
118By default the process command is interpreted as a Bash script. However, any other scripting language can be used just simply starting the script with the corresponding Shebang declaration. For example:
GROOVY
//process_python.nf
process PROCESS_READS {
script:
"""
#!/usr/bin/env python
import gzip
reads = 0
bases = 0
with gzip.open('${projectDir}/data/yeast/reads/ref1_1.fq.gz', 'rb') as read:
for id in read:
seq = next(read)
reads += 1
bases += len(seq.strip())
next(read)
next(read)
print("reads", reads)
print("bases", bases)
"""
}
workflow {
PROCESS_READS()
}OUTPUT
N E X T F L O W ~ version 24.04.4
Launching `process_python.nf` [mad_montalcini] DSL2 - revision: ee25d49465
executor > local (1)
[b4/a100c3] PROCESS_READS [100%] 1 of 1 ✔
reads 14677
bases 1482377This allows the use of a different programming languages which may better fit a particular job. However, for large chunks of code it is suggested to save them into separate files and invoke them from the process script.
Associated scripts
Scripts such as the one in the example below,
process_reads.py, can be stored in a bin
folder at the same directory level as the Nextflow workflow script that
invokes them, and given execute permission. Nextflow will automatically
add this folder to the PATH environment variable. To invoke
the script in a Nextflow process, simply use its filename on its own
rather than invoking the interpreter e.g. process_reads.py
instead of python process_reads.py. Note
The script process_reads.py must be executable to run.
PYTHON
#!/usr/bin/env python
# process_reads.py
import gzip
import sys
reads = 0
bases = 0
with gzip.open(sys.argv[1], 'rb') as read:
for id in read:
seq = next(read)
reads += 1
bases += len(seq.strip())
next(read)
next(read)
print("reads", reads)
print("bases", bases)GROOVY
//process_python_script.nf
process PROCESS_READS {
script:
"""
process_reads.py ${projectDir}/data/yeast/reads/ref1_1.fq.gz
"""
}
workflow {
PROCESS_READS()
}OUTPUT
N E X T F L O W ~ version 23.10.1
Launching `pr.nf` [kickass_legentil] DSL2 - revision: 3b9eee1d47
executor > local (1)
[88/759311] process > PROCESS_READS [100%] 1 of 1 ✔
reads 14677
bases 1482377Associated scripts
Scripts such as the one in the example above,
process_reads.py, can be stored in a bin
folder at the same directory level as the Nextflow workflow script that
invokes them, and given execute permission. Nextflow will automatically
add this folder to the PATH environment variable. To invoke
the script in a Nextflow process, simply use its filename on its own
rather than invoking the interpreter e.g. process_reads.py
instead of python process_reads.py.
Script parameters
The command in the script block can be defined
dynamically using Nextflow variables e.g. ${projectDir}. To
reference a variable in the script block you can use the $
in front of the Nextflow variable name, and additionally you can add
{} around the variable name
e.g. ${projectDir}.
Variable substitutions
Similar to bash scripting Nextflow uses the $ character
to introduce variable substitutions. The variable name to be expanded
may be enclosed in braces {variable_name}, which are
optional but serve to protect the variable to be expanded from
characters immediately following it which could be interpreted as part
of the name. It is a good rule of thumb to always use the
{} syntax because it enhances readability and clarity,
ensures correct variable interpretation, and prevents potential syntax
errors in complex expressions.
In the example below the variable chr is set to the
value A at the top of the Nextflow script. The variable is referenced
using the $chr syntax within the multi-line string
statement in the script block. A Nextflow variable can be
used multiple times in the script block.
GROOVY
//process_script.nf
chr = "A"
process CHR_COUNT {
script:
"""
printf "Number of sequences for chromosome ${chr} :"
zgrep -c '>Y'${chr} ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz
"""
}
workflow {
CHR_COUNT()
}In most cases we do not want to hard code parameter values. We saw in
the parameter episode the use of a special Nextflow variable
params that can be used to assign values from the command
line. You would do this by adding a key name to the params variable and
specifying a value, like params.keyname = value
In the example below we define the variable params.chr
with a default value of A in the Nextflow script.
GROOVY
//process_script_params.nf
params.chr = "A"
process CHR_COUNT {
script:
"""
printf 'Number of sequences for chromosome '${params.chr}':'
zgrep -c '^>Y'${params.chr} ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz
"""
}
workflow {
CHR_COUNT()
}Remember, we can change the default value of chr to a
different value such as B, by running the Nextflow script
using the command below. Note: parameters to the
workflow have two hyphens --.
OUTPUT
N E X T F L O W ~ version 24.04.3
Launching `process_script_params.nf` [pedantic_mandelbrot] DSL2 - revision: 538e3c2b38
executor > local (1)
[19/6d96a0] process > CHR_COUNT [100%] 1 of 1 ✔
Number of sequences for chromosome B:456Script parameters
For the Nextflow script below.
GROOVY
//process_exercise_script_params.nf
process COUNT_BASES {
script:
"""
zgrep -v '^>' ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz|grep -o A|wc -l
"""
}
workflow {
COUNT_BASES()
}Add a parameter params.base to the script and uses the
variable ${param.base} insides the script. Run the pipeline
using a base value of C using the --base
command line option.
Note: The Nextflow option
-process.debug will print the process’ stdout to the
terminal.
GROOVY
//process_exercise_script_params.nf
params.base='A'
process COUNT_BASES {
script:
"""
zgrep -v '^>' ${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz|grep -o ${params.base}|wc -l
"""
}
workflow {
COUNT_BASES()
}OUTPUT
N E X T F L O W ~ version 21.04.0
Launching `process_script_params.nf ` [nostalgic_jones] - revision: 9feb8de4fe
executor > local (1)
[92/cdc9de] process > COUNT_BASES [100%] 1 of 1 ✔
1677188Bash variables
Nextflow uses the same Bash syntax for variable substitutions,
$variable, in strings. However, Bash variables need to be
escaped using \ character in front of
\$variable name.
In the example below we will set a bash variable NUMIDS
then echo the value of NUMIDS in our script block.
GROOVY
//process_escape_bash.nf
process NUM_IDS {
script:
"""
#set bash variable NUMIDS
NUMIDS=`zgrep -c '^>' $params.transcriptome`
echo 'Number of sequences'
printf "%'d\n" \$NUMIDS
"""
}
params.transcriptome = "${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
workflow {
NUM_IDS()
}Shell
Another alternative is to use a shell block definition
instead of script. When using the shell
statement Bash variables are referenced in the normal way
$my_bash_variable; However, the shell
statement uses a different syntax for Nextflow variable substitutions:
!{nextflow_variable}, which is needed to use both Nextflow
and Bash variables in the same script.
For example in the script below that uses the shell
statement we reference the Nextflow variables as
!{projectDir} , and the Bash variable as
${NUMCHAR} and ${NUMLINES}.
GROOVY
//process_shell.nf
process NUM_IDS {
shell:
//Shell script definition requires the use of single-quote ' delimited strings
'''
#set bash variable NUMIDS
NUMIDS=`zgrep -c '^>' !{params.transcriptome}`
echo 'Number of sequences'
printf $NUMIDS
'''
}
params.transcriptome = "${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
workflow {
NUM_IDS()
}Conditional script execution
Sometimes you want to change how a process is run depending on some
condition. In Nextflow scripts we can use conditional statements such as
the if statement or any other expression evaluating to
boolean value true or false.
If statement
The if statement uses the same syntax common to other
programming languages such Java, C, JavaScript, etc.
GROOVY
if( < boolean expression > ) {
// true branch
}
else if ( < boolean expression > ) {
// true branch
}
else {
// false branch
}For example, the Nextflow script below will use the if
statement to change what the COUNT process counts depending on the
Nextflow variable params.method.
GROOVY
//process_conditional.nf
params.method = 'ids'
params.transcriptome = "$projectDir/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
process COUNT {
script:
if( params.method == 'ids' ) {
"""
echo Number of sequences in transciptome
zgrep -c "^>" $params.transcriptome
"""
}
else if( params.method == 'bases' ) {
"""
echo Number of bases in transciptome
zgrep -v "^>" $params.transcriptome|grep -o "."|wc -l
"""
}
else {
"""
echo Unknown method $params.method
"""
}
}
workflow {
COUNT()
}OUTPUT
N E X T F L O W ~ version 21.04.0
Launching `juggle_processes.nf` [cheeky_shirley] - revision: 588f20ae5a
[01/60b08d] process > COUNT [100%] 1 of 1 ✔
Number of sequences in transciptome
6612Inputs
Processes are isolated from each other but can communicate by sending
values and files via Nextflow channels from input and into
output blocks.
The input block defines which channels the process is
expecting to receive input from. The number of elements in input
channels determines the process dependencies and the number of times a
process executes.
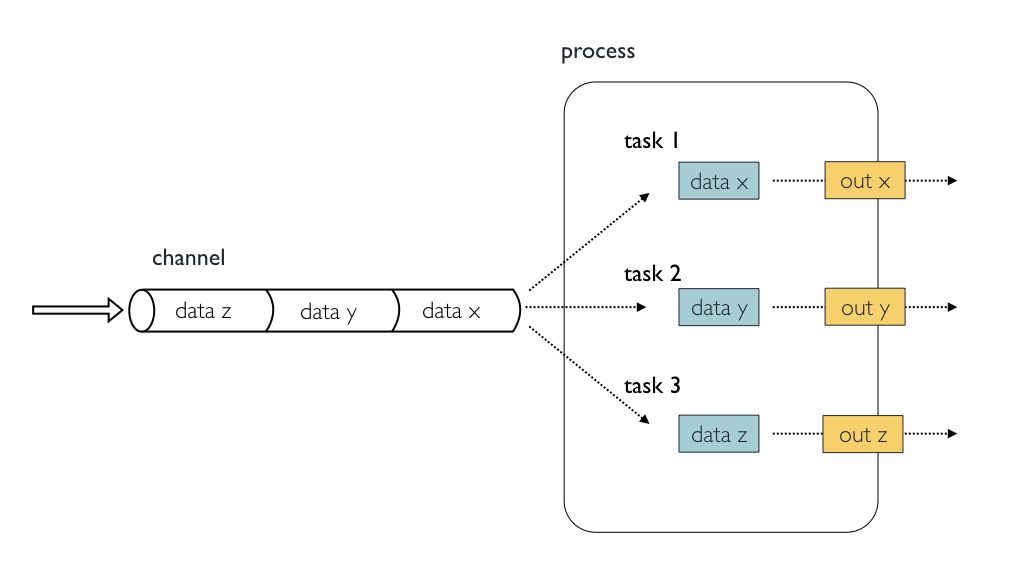
You can only define one input block at a time and it must contain one or more input declarations.
The input block follows the syntax shown below:
The input qualifier declares the type of data to be received.
Input qualifiers
-
val: Lets you access the received input value by its name as a variable in the process script. -
env: Lets you use the input value to set an environment variable named as the specified input name. -
path: Lets you handle the received value as a file, staging the file properly in the execution context. -
stdin: Lets you forward the received value to the process stdin special file. -
tuple: Lets you handle a group of input values having one of the above qualifiers. -
each: Lets you execute the process for each entry in the input collection. A complete list of inputs can be found here.
Input values
The val qualifier allows you to receive value data as
input. It can be accessed in the process script by using the specified
input name, as shown in the following example:
GROOVY
//process_input_value.nf
process PRINTCHR {
input:
val chr
script:
"""
echo processing chromosome $chr
"""
}
chr_ch = Channel.of( 'A' .. 'P' )
workflow {
PRINTCHR(chr_ch)
}OUTPUT
N E X T F L O W ~ version 21.04.0
Launching `process_input_value.nf` [wise_kalman] - revision: 7f90e1bfc5
executor > local (24)
[b1/88df3f] process > PRINTCHR (16) [100%] 24 of 24 ✔
processing chromosome C
processing chromosome L
processing chromosome A
..truncated...In the above example the process is executed 16 times; each time a
value is received from the queue channel chr_ch it is used
to run the process.
Channel order
The channel guarantees that items are delivered in the same order as they have been sent, but since the process is executed in a parallel manner, there is no guarantee that they are processed in the same order as they are received.
Input files
When you need to handle files as input, you need the
path qualifier. Using the path qualifier means
that Nextflow will stage it in the process execution directory, and it
can be accessed in the script by using the name specified in the input
declaration.
The input file name can be defined dynamically by defining the input
name as a Nextflow variable and referenced in the script using the
$variable_name syntax.
For example, in the script below, we assign the variable name
read to the input files using the path
qualifier. The file is referenced using the variable substitution syntax
${read} in the script block:
GROOVY
//process_input_file.nf
process NUMLINES {
input:
path read
script:
"""
printf '${read} '
gunzip -c ${read} | wc -l
"""
}
reads_ch = Channel.fromPath( 'data/yeast/reads/ref*.fq.gz' )
workflow {
NUMLINES(reads_ch)
}OUTPUT
[cd/77af6d] process > NUMLINES (1) [100%] 6 of 6 ✔
ref1_1.fq.gz 58708
ref3_2.fq.gz 52592
ref2_2.fq.gz 81720
ref2_1.fq.gz 81720
ref3_1.fq.gz 52592
ref1_2.fq.gz 58708Callout
The input name can also be defined as a user-specified filename
inside quotes. For example, in the script below, the name of the file is
specified as 'sample.fq.gz' in the input definition and can
be referenced by that name in the script block.
GROOVY
//process_input_file_02.nf
process NUMLINES {
input:
path 'sample.fq.gz'
script:
"""
printf 'sample.fq.gz '
gunzip -c sample.fq.gz | wc -l
"""
}
reads_ch = Channel.fromPath( 'data/yeast/reads/ref*.fq.gz' )
workflow {
NUMLINES(reads_ch)
}OUTPUT
[d2/eb0e9d] process > NUMLINES (1) [100%] 6 of 6 ✔
sample.fq.gz 58708
sample.fq.gz 52592
sample.fq.gz 81720
sample.fq.gz 81720
sample.fq.gz 52592
sample.fq.gz 58708File Objects as inputs
When a process declares an input file, the corresponding channel
elements must be file objects, i.e. created with the path helper
function from the file specific channel factories,
e.g. Channel.fromPath or
Channel.fromFilePairs.
Add input channel
For the script process_exercise_input.nf:
- Define a Channel using
fromPathfor the transcriptomeparams.transcriptome. - Add an input channel that takes the transcriptome channel as a file input.
- Replace
params.transcriptomein thescript:block with the input variable you defined in theinput:definition.
GROOVY
//process_exercise_input.nf
params.chr = "A"
params.transcriptome = "${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
process CHR_COUNT {
script:
"""
printf 'Number of sequences for chromosome '${params.chr}':'
zgrep -c '^>Y'${params.chr} ${params.transcriptome}
"""
}
workflow {
CHR_COUNT()
}Then run your script using
GROOVY
params.chr = "A"
params.transcriptome = "${projectDir}/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
process CHR_COUNT {
input:
path transcriptome
script:
"""
printf 'Number of sequences for chromosome '${params.chr}':'
zgrep -c '^>Y'${params.chr} ${transcriptome}
"""
}
transcriptome_ch = channel.fromPath(params.transcriptome)
workflow {
CHR_COUNT(transcriptome_ch)
}OUTPUT
N E X T F L O W ~ version 21.10.6
Launching `process_exercise_input.nf` [focused_jang] - revision: e32caf0dcb
executor > local (1)
[00/14ce67] process > CHR_COUNT (1) [100%] 1 of 1 ✔
Number of sequences for chromosome A:118Combining input channels
A key feature of processes is the ability to handle inputs from multiple channels. However, it’s important to understand how the number of items within the multiple channels affect the execution of a process.
Consider the following example:
GROOVY
//process_combine.nf
process COMBINE {
input:
val x
val y
script:
"""
echo $x and $y
"""
}
num_ch = Channel.of(1, 2, 3)
letters_ch = Channel.of('a', 'b', 'c')
workflow {
COMBINE(num_ch, letters_ch)
}Both channels contain three elements, therefore the process is executed three times, each time with a different pair:
OUTPUT
2 and b
1 and a
3 and cWhat is happening is that the process waits until it receives an input value from all the queue channels declared as input.
When this condition is verified, it uses up the input values coming from the respective queue channels, runs the task. This logic repeats until one or more queue channels have no more content. The process then stops.
What happens when not all channels have the same number of elements?
For example:
GROOVY
//process_combine_02.nf
process COMBINE {
input:
val x
val y
script:
"""
echo $x and $y
"""
}
ch_num = Channel.of(1, 2)
ch_letters = Channel.of('a', 'b', 'c', 'd')
workflow {
COMBINE(ch_num, ch_letters)
}In the above example the process is executed only two times, because when a queue channel has no more data to be processed it stops the process execution.
OUTPUT
2 and b
1 and aValue channels and process termination
Note however that value channels,
Channel.value, do not affect the process termination.
To better understand this behaviour compare the previous example with the following one:
GROOVY
//process_combine_03.nf
process COMBINE {
input:
val x
val y
script:
"""
echo $x and $y
"""
}
ch_num = Channel.value(1)
ch_letters = Channel.of('a', 'b', 'c')
workflow {
COMBINE(ch_num, ch_letters)
}In this example the process is run three times.
OUTPUT
1 and b
1 and a
1 and cCombining input channels
GROOVY
// process_exercise_combine_answer.nf
process COMBINE {
input:
path transcriptome
val chr
script:
"""
zgrep -c ">Y${chr}" ${transcriptome}
"""
}
transcriptome_ch = channel.fromPath('data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz', checkIfExists: true)
chr_ch = channel.of("A")
workflow {
COMBINE(transcriptome_ch, chr_ch)
}OUTPUT
N E X T F L O W ~ version 24.04.4
Launching `process_exercise_combine.nf` [fabulous_kare] DSL2 - revision: 1eade0a2e9
executor > local (1)
[e0/b05fe7] COMBINE (1) [100%] 1 of 1 ✔
118Input repeaters
We saw previously that by default the number of times a process runs
is defined by the queue channel with the fewest items. However, the
each qualifier allows you to repeat the execution of a
process for each item in a list or a queue channel, every time new data
is received.
For example if we can fix the previous example by using the input
qualifer each for the letters queue channel:
GROOVY
//process_repeat.nf
process COMBINE {
input:
val x
each y
script:
"""
echo $x and $y
"""
}
ch_num = Channel.of(1, 2)
ch_letters = Channel.of('a', 'b', 'c', 'd')
workflow {
COMBINE(ch_num, ch_letters)
}The process will run eight times.
OUTPUT
2 and d
1 and a
1 and c
2 and b
2 and c
1 and d
1 and b
2 and aInput repeaters
Extend the script process_exercise_repeat.nf by adding
more values to the chr queue channel e.g. A to P and
running the process for each value.
GROOVY
//process_exercise_repeat.nf
process COMBINE {
input:
path transcriptome
val chr
script:
"""
printf "Number of sequences for chromosome $chr: "
zgrep -c "^>Y${chr}" ${transcriptome}
"""
}
transcriptome_ch = channel.fromPath('data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz', checkIfExists: true)
chr_ch = channel.of('A')
workflow {
COMBINE(transcriptome_ch, chr_ch)
}How many times does this process run?
GROOVY
//process_exercise_repeat_answer.nf
process COMBINE {
input:
path transcriptome
each chr
script:
"""
printf "Number of sequences for chromosome $chr: "
zgrep -c "^>Y${chr}" ${transcriptome}
"""
}
transcriptome_ch = channel.fromPath('data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz', checkIfExists: true)
chr_ch = channel.of('A'..'P')
workflow {
COMBINE(transcriptome_ch, chr_ch)
}Then run the script.
This process runs 16 times.
OUTPUT
N E X T F L O W ~ version 24.04.4
Launching `process_exercise_repeat.nf` [ecstatic_turing] DSL2 - revision: 17891a7528
executor > local (16)
[65/389033] COMBINE (13) [ 62%] 10 of 16
executor > local (16)
[6d/f803e5] COMBINE (9) [100%] 16 of 16 ✔
Number of sequences for chromosome J: 398
Number of sequences for chromosome G: 583
Number of sequences for chromosome O: 597
Number of sequences for chromosome N: 435
Number of sequences for chromosome B: 456
Number of sequences for chromosome E: 323
executor > local (16)
[6d/f803e5] COMBINE (9) [100%] 16 of 16 ✔
Number of sequences for chromosome J: 398
Number of sequences for chromosome G: 583
Number of sequences for chromosome O: 597
Number of sequences for chromosome N: 435
Number of sequences for chromosome B: 456
Number of sequences for chromosome E: 323
Number of sequences for chromosome K: 348
Number of sequences for chromosome H: 321
Number of sequences for chromosome C: 186
Number of sequences for chromosome M: 505
Number of sequences for chromosome L: 580
Number of sequences for chromosome A: 118
Number of sequences for chromosome D: 836
Number of sequences for chromosome F: 140
Number of sequences for chromosome P: 513
Number of sequences for chromosome I: 245Key Points
- A Nextflow process is an independent step in a workflow.
- Processes contain up to five definition blocks including: directives, inputs, outputs, when clause and finally a script block.
- The script block contains the commands you would like to run.
- A process should have a script but the other four blocks are optional.
- Inputs are defined in the input block with a type qualifier and a name.