Getting Started with Nextflow
Last updated on 2024-06-10 | Edit this page
Overview
Questions
- What is a workflow and what are workflow management systems?
- Why should I use a workflow management system?
- What is Nextflow?
- What are the main features of Nextflow?
- What are the main components of a Nextflow script?
- How do I run a Nextflow script?
Objectives
- Understand what a workflow management system is.
- Understand the benefits of using a workflow management system.
- Explain the benefits of using Nextflow as part of your bioinformatics workflow.
- Explain the components of a Nextflow script.
- Run a Nextflow script.
Workflows
Analysing data involves a sequence of tasks, including gathering, cleaning, and processing data. This sequence of tasks is called a workflow or a pipeline. These workflows typically require executing multiple software packages, sometimes running on different computing environments, such as a desktop or a compute cluster. Traditionally these workflows have been joined together in scripts using general purpose programming languages such as Bash or Python.
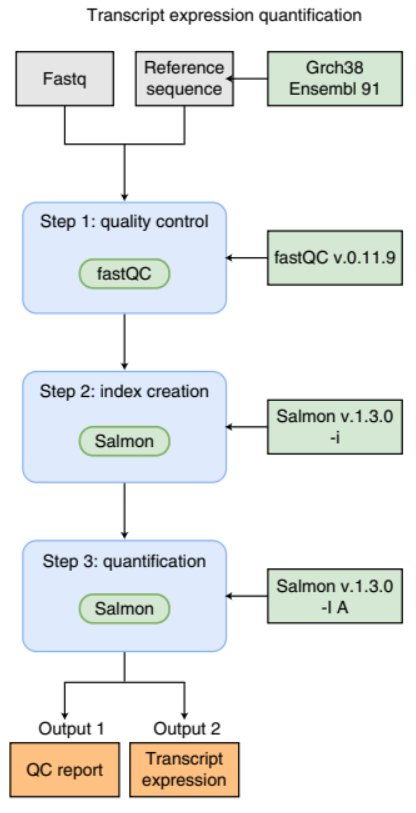
An example of a simple bioinformatics RNA-Seq pipeline.
However, as workflows become larger and more complex, the management of the programming logic and software becomes difficult.
Workflow management systems
Workflow Management Systems (WfMS) such as Snakemake, Galaxy, and Nextflow have been developed specifically to manage computational data-analysis workflows in fields such as bioinformatics, imaging, physics, and chemistry. These systems contain multiple features that simplify the development, monitoring, execution and sharing of pipelines, such as:
- Run time management
- Software management
- Portability & Interoperability
- Reproducibility
- Re-entrancy
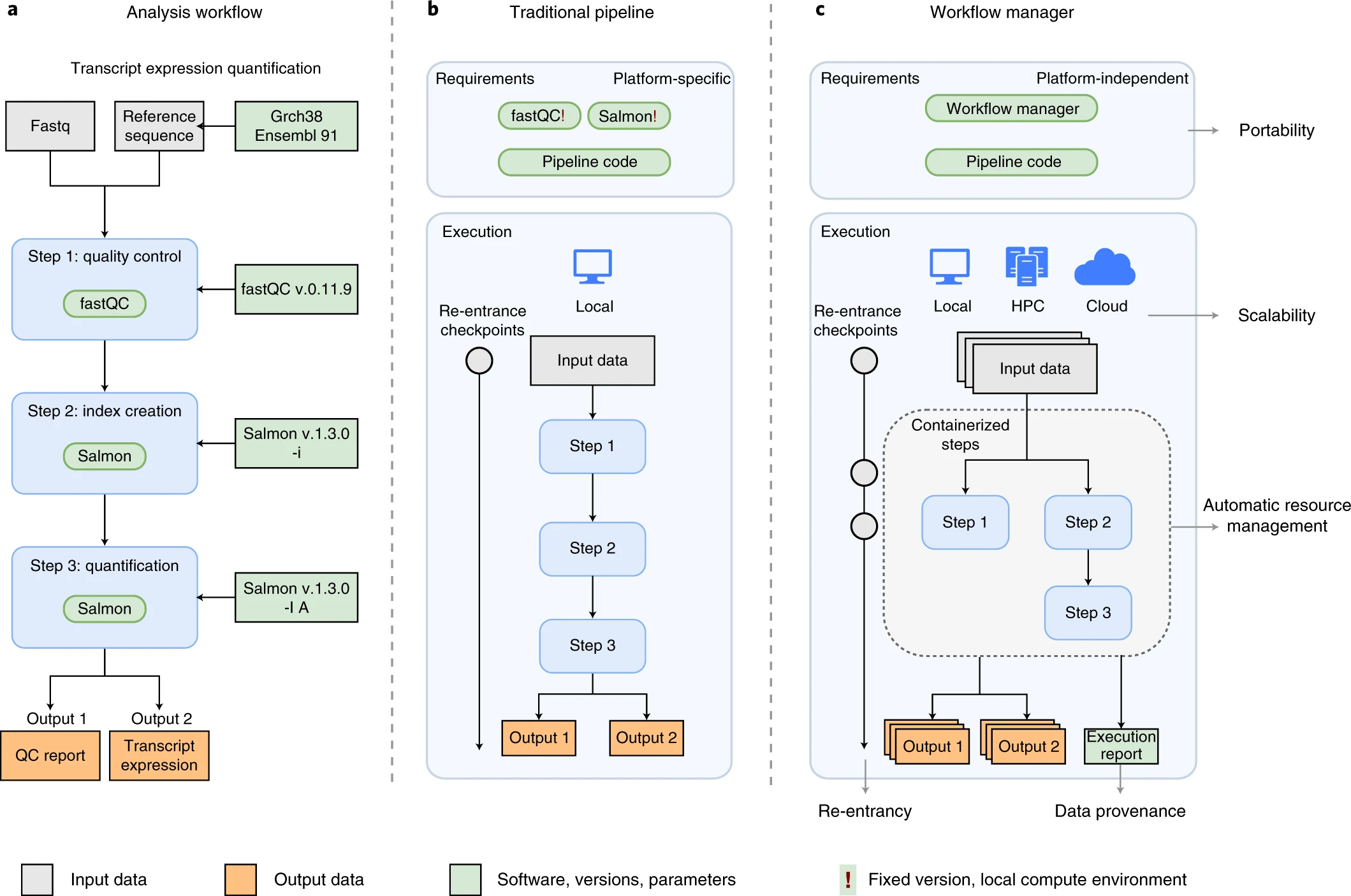
An example of differences between running a specific analysis workflow using a traditional pipeline or a WfMS-based pipeline. Source: Wratten, L., Wilm, A. & Göke, J. Reproducible, scalable, and shareable analysis pipelines with bioinformatics workflow managers. Nat Methods 18, 1161–1168 (2021). https://doi.org/10.1038/s41592-021-01254-9
Nextflow core features
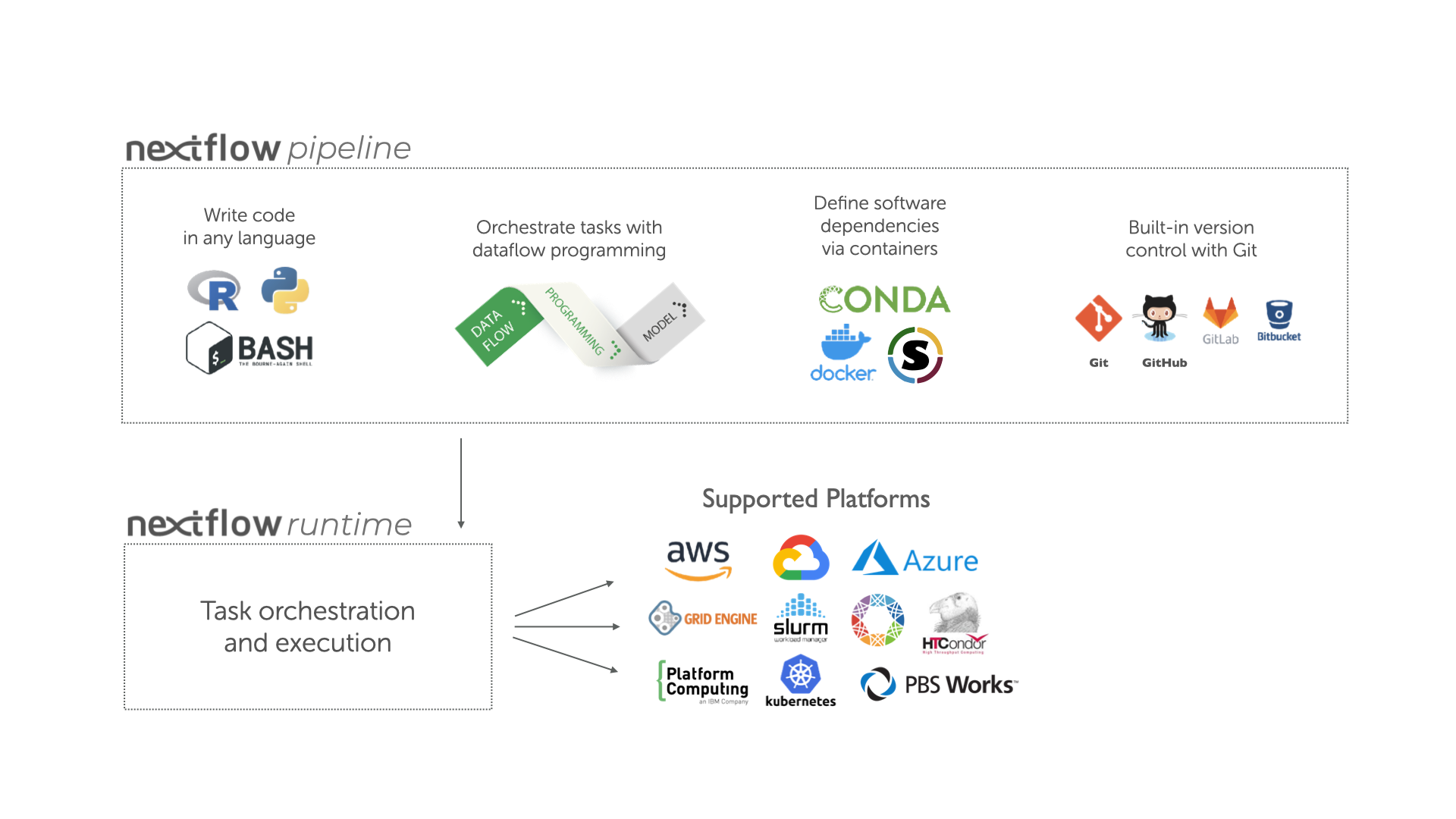
Overview of Nextflow core features.
Fast prototyping: A simple syntax for writing pipelines that enables you to reuse existing scripts and tools for fast prototyping.
Reproducibility: Nextflow supports several container technologies, such as Docker and Singularity, as well as the package manager Conda. This, along with the integration of the GitHub code sharing platform, allows you to write self-contained pipelines, manage versions and to reproduce any previous result when re-run, including on different computing platforms.
Portability & interoperability: Nextflow’s syntax separates the functional logic (the steps of the workflow) from the execution settings (how the workflow is executed). This allows the pipeline to be run on multiple platforms, e.g. local compute vs. a university compute cluster or a cloud service like AWS, without changing the steps of the workflow.
Simple parallelism: Nextflow is based on the dataflow programming model which greatly simplifies the splitting of tasks that can be run at the same time (parallelisation).
Continuous checkpoints & re-entrancy: All the intermediate results produced during the pipeline execution are automatically tracked. This allows you to resume its execution from the last successfully executed step, no matter what the reason was for it stopping.
Processes, channels, and workflows
Nextflow workflows have three main parts: processes, channels, and workflows.
Processes describe a task to be run. A process script can be written in any scripting language that can be executed by the Linux platform (Bash, Perl, Ruby, Python, R, etc.). Processes spawn a task for each complete input set. Each task is executed independently and cannot interact with other tasks. The only way data can be passed between process tasks is via asynchronous queues, called channels.
Processes define inputs and outputs for a task. Channels are then used to manipulate the flow of data from one process to the next.
The interaction between processes, and ultimately the pipeline execution flow itself, is then explicitly defined in a workflow section.
In the following example we have a channel containing three elements, e.g., three data files. We have a process that takes the channel as input. Since the channel has three elements, three independent instances (tasks) of that process are run in parallel. Each task generates an output, which is passed to another channel and used as input for the next process.
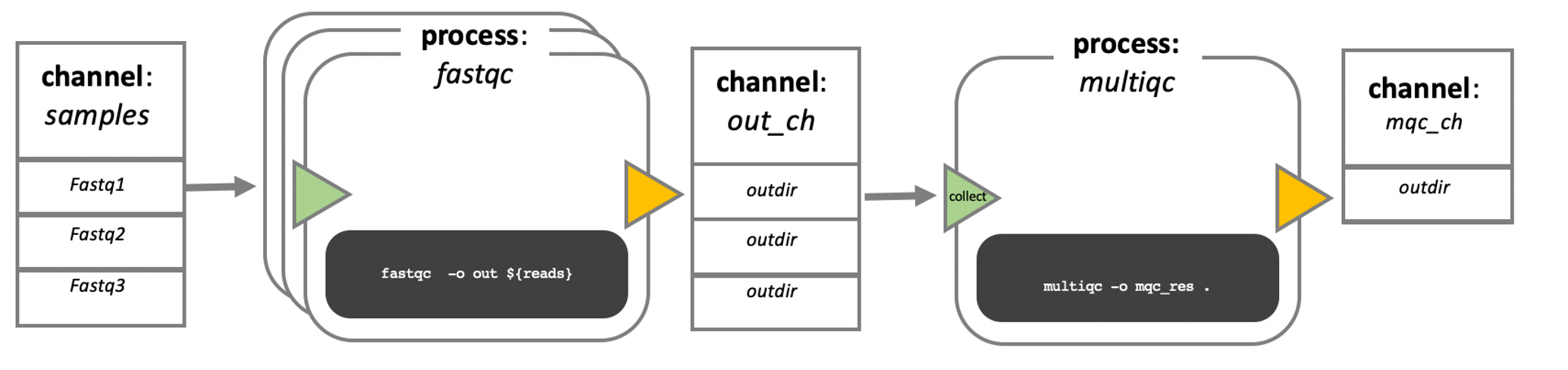
Nextflow process flow diagram.
Workflow execution
While a process defines what command or script has to be
executed, the executor determines how that script is
actually run in the target system.
If not otherwise specified, processes are executed on the local computer. The local executor is very useful for pipeline development, testing, and small-scale workflows, but for large-scale computational pipelines, a High Performance Cluster (HPC) or Cloud platform is often required.
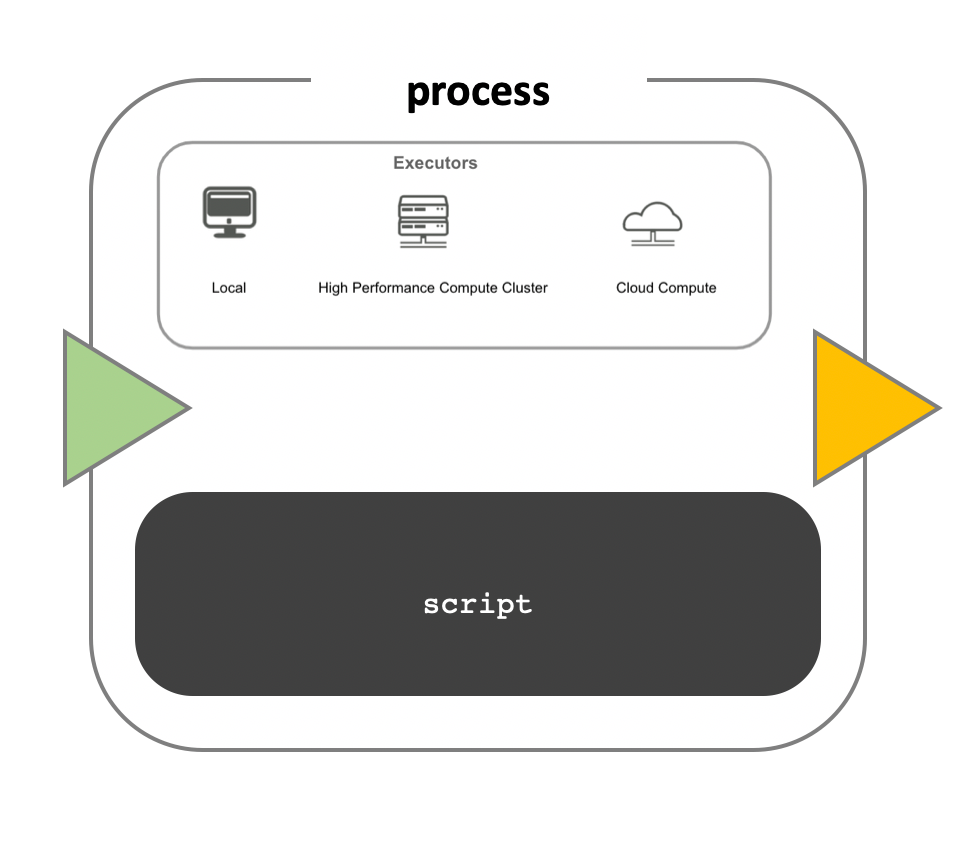
Nextflow Executors
Nextflow provides a separation between the pipeline’s functional logic and the underlying execution platform. This makes it possible to write a pipeline once, and then run it on your computer, compute cluster, or the cloud, without modifying the workflow, by defining the target execution platform in a configuration file.
Nextflow provides out-of-the-box support for major batch schedulers and cloud platforms such as Sun Grid Engine, SLURM job scheduler, AWS Batch service and Kubernetes; a full list can be found here.
Your first script
We are now going to look at a sample Nextflow script that counts the
number of lines in a file. Create the file word_count.nf in
the current directory using your favourite text editor and copy-paste
the following code:
GROOVY
#!/usr/bin/env nextflow
/*
========================================================================================
Workflow parameters are written as params.<parameter>
and can be initialised using the `=` operator.
========================================================================================
*/
params.input = "data/yeast/reads/ref1_1.fq.gz"
/*
========================================================================================
Input data is received through channels
========================================================================================
*/
input_ch = Channel.fromPath(params.input)
/*
========================================================================================
Main Workflow
========================================================================================
*/
workflow {
// The script to execute is called by it's process name, and input is provided between brackets.
NUM_LINES(input_ch)
/* Process output is accessed using the `out` channel.
The channel operator view() is used to print process output to the terminal. */
NUM_LINES.out.view()
}
/*
========================================================================================
A Nextflow process block. Process names are written, by convention, in uppercase.
This convention is used to enhance workflow readability.
========================================================================================
*/
process NUM_LINES {
input:
path read
output:
stdout
script:
"""
# Print file name
printf '${read}\\t'
# Unzip file and count number of lines
gunzip -c ${read} | wc -l
"""
}This is a Nextflow script, which contains the following:
- An optional interpreter directive (“Shebang”) line, specifying the location of the Nextflow interpreter.
- A multi-line Nextflow comment, written using C style block comments, there are more comments later in the file.
- A pipeline parameter
params.inputwhich is given a default value, of the relative path to the location of a compressed fastq file, as a string. - A Nextflow channel
input_chused to read in data to the workflow. - An unnamed
workflowexecution block, which is the default workflow to run. - A call to the process
NUM_LINES. - An operation on the process output, using the channel operator
.view(). - A Nextflow process block named
NUM_LINES, which defines what the process does. - An
inputdefinition block that assigns theinputto the variableread, and declares that it should be interpreted as a file path. - An
outputdefinition block that uses the Linux/Unix standard output streamstdoutfrom the script block. - A script block that contains the bash commands
printf '${read}'andgunzip -c ${read} | wc -l.
Running Nextflow scripts
To run a Nextflow script use the command
nextflow run <script_name>.
You should see output similar to the text shown below:
OUTPUT
N E X T F L O W ~ version 21.04.3
Launching `word_count.nf` [fervent_babbage] - revision: c54a707593
executor > local (1)
[21/b259be] process > NUM_LINES (1) [100%] 1 of 1 ✔
ref1_1.fq.gz 58708- The first line shows the Nextflow version number.
- The second line shows the run name
fervent_babbage(adjective and scientist name) and revision idc54a707593. - The third line tells you the process has been executed locally
(
executor > local). - The next line shows the process id
21/b259be, process name, number of cpus, percentage task completion, and how many instances of the process have been run. - The final line is the output of the
.view()operator.
Quick recap
- A workflow is a sequence of tasks that process a set of data, and a workflow management system (WfMS) is a computational platform that provides an infrastructure for the set-up, execution and monitoring of workflows.
- Nextflow scripts comprise of channels for controlling inputs and outputs, and processes for defining workflow tasks.
- You run a Nextflow script using the
nextflow runcommand.
Key Points
- A workflow is a sequence of tasks that process a set of data.
- A workflow management system (WfMS) is a computational platform that provides an infrastructure for the set-up, execution and monitoring of workflows.
- Nextflow is a workflow management system that comprises both a runtime environment and a domain specific language (DSL).
- Nextflow scripts comprise of channels for controlling inputs and outputs, and processes for defining workflow tasks.
- You run a Nextflow script using the
nextflow runcommand.