Data Collection: ArrayExpress
Last updated on 2024-05-14 | Edit this page
Overview
Questions
- What format are processed RNA-Seq dataset stored on Array Express?
- How do I search for a dataset that meets my requirements on Array Express?
- Which files do I need to download?
Objectives
- Explain the expected data and file formats for processed RNA-Seq datasets on ArrayExpress
- Demonstrate ability to correctly identify the subset of files required to create a dataset for a simple supervised machine learning task (e.g., binary classification)
- Demonstrate ability to download and save processed RNA-Seq data files from ArrayExpress suitable for a simple supervised machine learning task (e.g., binary classification)
ArrayExpress File Formats
Array Express data sets comprise of four important file types that contain all of the information about an experiment:
| File Type | What It Is |
|---|---|
| Sample and Data Relationship Format (SDRF) | Tab-delimited file containing information about the samples, such as phenotypical information, and factors that may be of interest in a classification task. This file may also contain information about the relationships between the samples, and details of the file names of the raw data fastq for each sample. |
| Investigation Description Format (IDF) | Tab-delimited file containing top level information about the experiment including title, description, submitter contact details and protocols. |
| Raw data files | Sequence data and quality scores, typically stored in FASTQ format. There is however inconsistency - in some data sets, the term raw data refers to processed data in the form of raw counts data, meaning RNA-Seq counts that have not undergone further processing. |
| Processed data files | Processed data which includes raw counts or further processed abundance measures such as TPM and FPKM. The data may be provided as a separate file for each sample, or as a matrix of all samples |
Detailed information about BioStudies file formats is available in the MAGE-TAB Specification document.
Searching for a Dataset on ArrayExpress
We will use ArrayExpress to select an RNA-Seq dataset with a case control design, suitable for constructing a machine learning classification model. Machine learning based analyses generally perform better the larger the sample size, and may perform very poorly and give misleading results with insufficient samples. Given that, we’ll look for a datasets with a relatively large number of samples.
Let’s begin on the ArrayExpress home page.
We’ll apply the following filters to refine the results set, leaving all other filters as the default with no selection:
| Filter | Selection |
|---|---|
| Study type | rna-seq of coding rna |
| Organism | homo sapiens |
| Technology | sequencing assay |
| Assay by Molecule | rna assay |
| Processed Data Available | yes (check box) |
Now use the sort in the top right of the screen to sort the results by “Samples”, in descending order. The results should look similar to this:
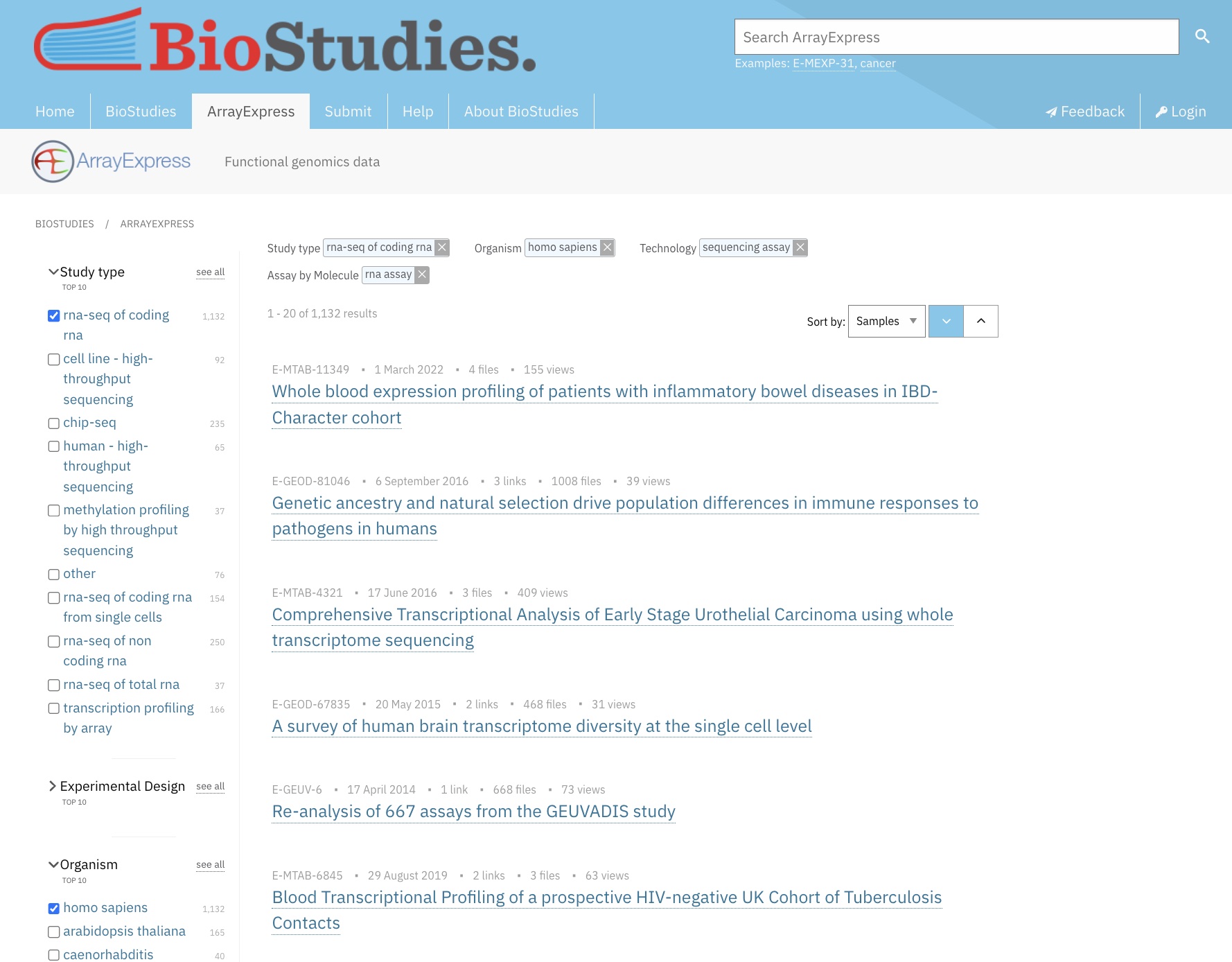
Illustrative Dataset: IBD Dataset
In the search results displayed above, the top-ranked dataset is E-MTAB-11349: Whole blood expression profiling of patients with inflammatory bowel diseases in the IBD-Character cohort. We’ll call it the IBD dataset.
The IBD dataset comprises human samples of patients with inflammatory bowel diseases and controls. The ‘Protocols’ section explains the main steps in the generation of the data, from RNA extraction and sample preparation, to sequencing and processing of the raw RNA-Seq data. The nucleic acid sequencing protocol gives details of the sequencing platform used to generate the raw fastq data, and the version of the human genome used in the alignment step to generate the count data. The normalization data transformation protocol gives the tools used to normalise raw counts data for sequence depth and sample composition, in this case normalisation is conducted using the R package DESeq2. DESEq2 contains a range of functions for the transformation and analysis of RNA-Seq data. Let’s look at some of the basic information on this dataset:
| Data Field | Values |
|---|---|
| Sample count | 590 |
| Experimental Design | case control design |
| Experimental factors | Normal, Ulcerative colitis, Crohn’s disease |
The dataset contains two alternative sets of processed data, a matrix of raw counts, and a matrix of processed counts that have been normalised using DESeq2, and filtered to only include transcripts with a mean raw expression read count > 10.
Downloading and Reading into R
Let’s download the SDRF file and the raw counts matrix - these are
the two files that contain the information we will need to build a
machine-learning classification model. In the data files box to the
right-hand side, check the following two files
E-MTAB-11349.sdrf.txt and
ArrayExpress-raw.csv, and save them in a folder called
data in the working directory for your R project. For
consistency, rename ArrayExpress-raw.csv as
E-MTAB-11349.counts.matrix.csv.
For convenience, a copy of the files is also stored on zenodo. You
can run the following code that uses the function
download.file() to download the files and save them
directly into your data directory. (You need to have
created the data directory beforehand).
To get or set a working directory in R getwd function
can be used to return an absolute file path representing the current
working directory of the R process; if not correct setwd
can be used to set the working directory to the desired location.
R
download.file(url = "https://zenodo.org/record/8125141/files/E-MTAB-11349.sdrf.txt",
destfile = "data/E-MTAB-11349.sdrf.txt")
download.file(url = "https://zenodo.org/record/8125141/files/E-MTAB-11349.counts.matrix.csv",
destfile = "data/E-MTAB-11349.counts.matrix.csv")
Raw counts matrix
In R studio, open your project workbook and read the raw counts data. Then check the dimensions of the matrix to confirm we have the expected number of samples and transcript IDs.
R
raw.counts.ibd <- read.table(file="data/E-MTAB-11349.counts.matrix.csv",
sep=",",
header=T,
fill=T,
check.names=F)
writeLines(sprintf("%i %s", c(dim(raw.counts.ibd)[1], dim(raw.counts.ibd)[2]), c("rows corresponding to transcript IDs", "columns corresponding to samples")))
OUTPUT
22751 rows corresponding to transcript IDs
592 columns corresponding to samplesR
raw.counts.ibd[1:10,1:8]
OUTPUT
read Sample 1 Sample 2 Sample 3 Sample 4 Sample 5 Sample 6
1 1 * 13961 16595 20722 17696 25703 20848
2 2 ERCC-00002 0 0 0 0 0 0
3 3 ERCC-00003 0 0 0 0 0 0
4 4 ERCC-00004 0 0 0 0 3 0
5 5 ERCC-00009 0 0 0 0 0 0
6 6 ERCC-00012 0 0 0 0 0 0
7 7 ERCC-00013 0 0 0 0 0 0
8 8 ERCC-00014 0 0 0 0 0 0
9 9 ERCC-00016 0 0 0 0 0 0
10 10 ERCC-00017 2 0 0 0 1 0SDRF File
Now let’s read the sdrf file (a plain text file) into R and check the dimensions of the file.
R
# read in the sdrf file
samp.info.ibd <- read.table(file="data/E-MTAB-11349.sdrf.txt", sep="\t", header=T, fill=T, check.names=F)
sprintf("There are %i rows, corresponding to the samples", dim(samp.info.ibd)[1])
OUTPUT
[1] "There are 590 rows, corresponding to the samples"R
sprintf("There are %i columns, corresponding to the available variables for each sample", dim(samp.info.ibd)[2])
OUTPUT
[1] "There are 32 columns, corresponding to the available variables for each sample"If we view the column names, we can see that the file does indeed contain a set of variables describing both phenotypical and experimental protocol information relating to each sample.
R
colnames(samp.info.ibd)
OUTPUT
[1] "Source Name"
[2] "Characteristics[organism]"
[3] "Characteristics[age]"
[4] "Unit[time unit]"
[5] "Term Source REF"
[6] "Term Accession Number"
[7] "Characteristics[developmental stage]"
[8] "Characteristics[sex]"
[9] "Characteristics[individual]"
[10] "Characteristics[disease]"
[11] "Characteristics[organism part]"
[12] "Material Type"
[13] "Protocol REF"
[14] "Protocol REF"
[15] "Protocol REF"
[16] "Extract Name"
[17] "Material Type"
[18] "Comment[LIBRARY_LAYOUT]"
[19] "Comment[LIBRARY_SELECTION]"
[20] "Comment[LIBRARY_SOURCE]"
[21] "Comment[LIBRARY_STRATEGY]"
[22] "Protocol REF"
[23] "Performer"
[24] "Assay Name"
[25] "Technology Type"
[26] "Protocol REF"
[27] "Derived Array Data File"
[28] "Comment [Derived ArrayExpress FTP file]"
[29] "Protocol REF"
[30] "Derived Array Data File"
[31] "Comment [Derived ArrayExpress FTP file]"
[32] "Factor Value[disease]" Key Points
- ArrayExpress stores two standard files with information about each experiment: 1. Sample and Data Relationship Format (SDRF) and 2. Investigation Description Format (IDF).
- ArrayExpress provides raw and processed data for RNA-Seq datasets, typically stored as csv, tsv, or txt files.
- The filters on the ArrayExpress website allow you to select and download a dataset that suit your task.