Introduction
Overview
Teaching: 0 min
Exercises: 0 minQuestions
What are computational workflows, and why are they useful?
What is Common Workflow Language?
How are CWL workflows written?
How do CWL workflows compare to shell workflows?
What are the advantages of using CWL workflows?
Objectives
Recognise the advantages of using a computational workflow
Understand why you might use CWL instead of a shell script
Computational Workflows
A computational workflow is a formalised chain of software tools, which explicitly defines how data is input, how it flows between tools and how it is output. Computational workflows are widely used for data analysis and enable rapid innovation and decision making.
The most important benefit of computational workflows is that they require a user to write down, fully formalise and automate their dataflow and process. While this can be a challenge, it allows greater repeatability, shareability and robustness to be achieved.
Below is an image showing a graphical representation of a dummy workflow. You can see inputs being made on the left, data flowing and being processed by tools and passed to the output.
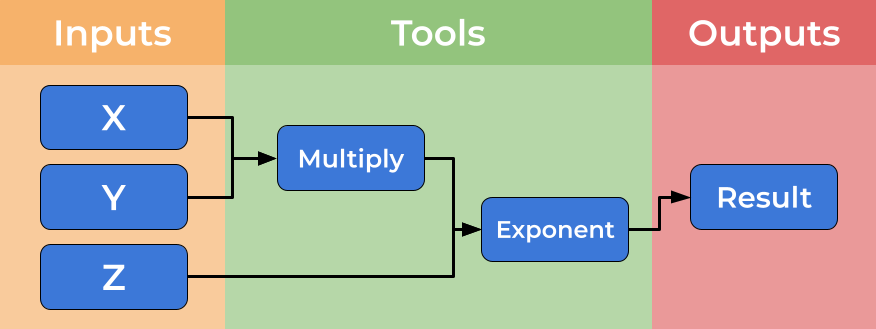
While you can imagine creating shell scripts (Bash or Make) to meet this need, using a formal workflow language (such as CWL) brings several further benefits such as introducing abstraction and improved scalability and portability. We will discuss some of these benefits here.
Computational workflows explicitly create a divide between a user’s dataflow and the computational details which underpin the chain of tools, placing these elements in separate files. The dataflow is described by the workflow, where tools are described at a high level. The tools implementation is specified by tool descriptors where the full complexity of a tools usage is handled. The image below shows how tool descriptors underpin workflow steps and hide complexity.
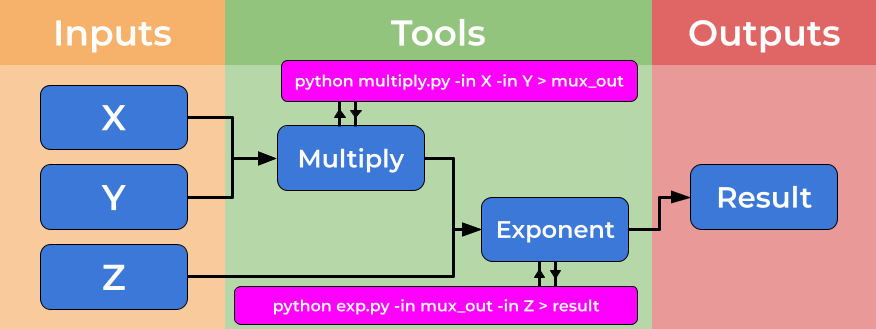
This abstraction allows the use of heterogeneous tools, potentially shared by third parties, and allows workflow users to connect and utilise a wide range of tools and techniques without the need for significant computational experience. An example of the strength of this approach is the ability for workflows to use (e.g. Docker) containers “under the hood” without the user needing to install, download or learn any further technologies.
By adapting tool descriptors to multiple platforms, this abstraction allows the same workflow to be used on different platforms, transparently to the workflow user. This means users can move between local development and cloud and HPC solutions seamlessly.
Computational workflow managers further extend this abstraction, providing high level tools for managing data and tools, aiming to help users to design and run computational workflows more easily. A computational workflow engine provides an interface for launching workflows, specifying and handling inputs and collecting and exporting outputs, they can also help users by storing completed steps of workflows, allowing workflows to be resumed part way, or rerun with minimal changes.
Further, computational workflow managers aid users with the automation, monitoring and provenance tracking of the dataflow. They may also help users to produce and understand reports and outputs from their workflow.
Another advantage of workflows is that by producing computational workflows in a standard format, and publishing them (alongside any data) with open access, allow dataflows to be shared in a more FAIR (Findable, Accessible, Interoperable, and Reusable) manner.
However, the rise in popularity of workflows has been matched by a rise in the number of disparate workflow managers that are available, each with their own syntax or methods for describing the tools and workflows, reducing portability and interoperability of these workflows. The Common Workflow Language (CWL) standard has been developed to address these problems, and to serve the general computational workflow needs described above.
In summary, computational workflows bring many benefits and an ideal computation workflow adopts and provides the properties below:
Handy Properties of Computational Workflows1
 |
Composition & Abstraction Using the best code written by 3rd parties Handle heterogeneity Shield Complexity & incompatibility Shareable reusable, re-mixable methods |
 |
Sharing & Adaptability Shared method, publishable know-how BYOD / parameters Different implementations Changes in execution infrastructure |
 |
Automation Repetitive reproducible pipelines Simulation sweeps Manage data and control flow Optimised monitoring & recovery Automated deployment |
 |
Reporting & Accreditation Provenance logging & data lineage Auto-documentation Result comparison |
 |
Scalability & Infrastructure Access Accessing infrastructures, datasets and tools Optimised computation and data handling Parallelisation Secure sensitive data access & management Interoperating datasets & permission handling |
 |
Portability Dependency handling Containerisation & packaging Moving between on premise & cloud |
Common Workflow Language
CWL is a free and open standard for describing command-line tool based workflows2. These standards provide a common, but reduced, set of abstractions that are both used in practice and implemented in many popular workflow managers. The CWL language is declarative, enabling computational workflows to be constructed from diverse software tools, executing each through their command-line interface.
Previously researchers might write shell scripts to link together these command-line tools. Although these scripts might provide a direct means of accessing the tools, writing and maintaining them requires specific knowledge of the system that they will be used on. Shell scripts are not easily portable, and so researchers can easily end up spending more time maintaining the scripts than carrying out their research. The aim of CWL is to reduce that barrier of usage of these tools to researchers.
CWL workflows are written in a subset of YAML, with a syntax that does not restrict the amount of detail provided for a tool or workflow. The execution model is explicit, all required elements of a tool’s runtime environment must be specified by the CWL tool-description author. On top of these basic requirements they can also add hints or requirements to the tool-description, helping to guide users (and workflow engines) on what resources are needed for a tool.
The CWL standards explicitly support the use of software container technologies, helping ensure that the execution of tools is reproducible. Data locations are explicitly defined, and working directories kept separate for each tool invocation. These standards ensure the portability of tools and workflows, allowing the same workflows to be run on your local machine, or in a HPC or cloud environment, with minimal changes required.
RNA sequencing example
In this tutorial a bio-informatics RNA-sequencing analysis is used as an example. However, there is no specific knowledge needed for this tutorial. RNA-sequencing is a technique which examines the quantity and sequences of RNA in a sample using next-generation sequencing. The RNA reads are analyzed to measure the relative numbers of different RNA molecules in the sample. This analysis is differential gene expression.
The process looks like this:
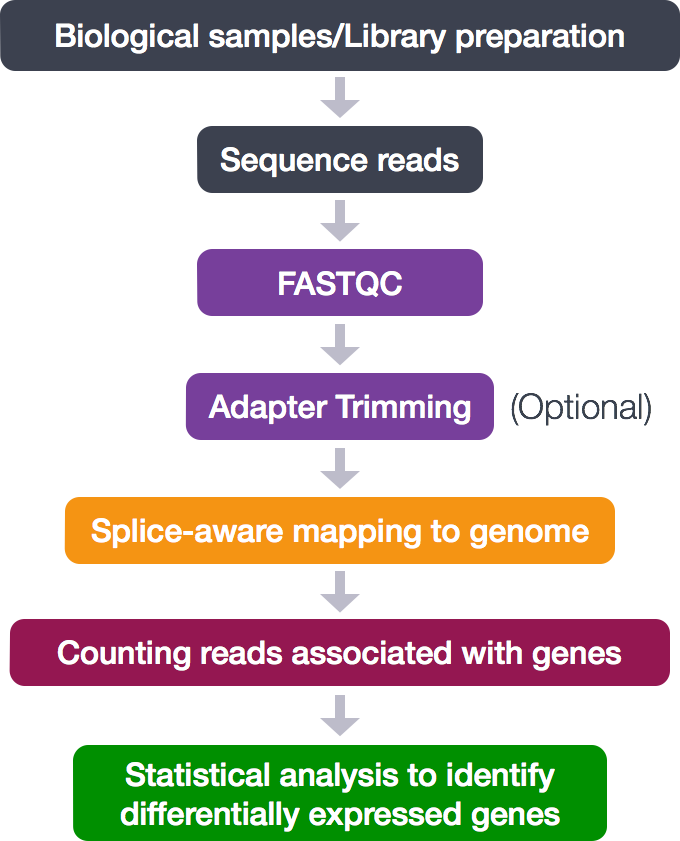
During this tutorial, only the middle analytical steps will be performed. The adapter trimming is skipped. These steps will be done:
- Quality control (FASTQC)
- Alignment (mapping)
- Counting reads associated with genes
The different tools necessary for this analysis are already available. In this tutorial a workflow will be set up to connect these tools and generate the desired output files.
-
C. Goble (2021): FAIR Computational Workflows. JOBIM Proceedings. https://www.slideshare.net/carolegoble/fair-computational-workflows-249721518 ↩
-
M. Wilkinson, M. Dumontier, I. Aalbersberg, et al. (2016): The FAIR Guiding Principles for scientific data management and stewardship. Scientific Data. https://doi.org/10.1038/sdata.2016.18 ↩
Key Points
CWL is a standard for describing workflows based on command-line tools
CWL workflows are written in a subset of YAML
A CWL workflow is more portable than a shell script
CWL supports software containers, supporting reproducibility on different machines
CWL and Shell Tools
Overview
Teaching: 0 min
Exercises: 0 minQuestions
What is the difference between a CWL tool description and a CWL workflow?
How can we create a tool descriptor?
How can we use this in a single step workflow?
Objectives
describe the relationship between a tool and its corresponding CWL document
exercise good practices when naming inputs and outputs
understand how to reference files for input and output
explain that only files explicitly mentioned in a description will be included in the output of a step/workflow
implement bulk capturing of all files produced by a step/workflow for debugging purposes
use STDIN and STDOUT as input and output
capture output written to a specific directory, the working directory, or the same directory where input is located
CWL workflows are written in the YAML syntax. This short tutorial explains the parts of YAML used in CWL. A CWL document contains the workflow and the requirements for running that workflow. All CWL documents should start with two lines of code:
cwlVersion: v1.2
class:
The cwlVersion string defines which standard of the language is required for the tool or workflow. The most recent version is v1.2.
The class field defines what this particular document is. The majority of CWL documents will fall into
one of two classes: CommandLineTool, or Workflow. The CommandLineTool class is used for
describing the interface for a command-line tool, while the Workflow class is used for connecting those
tool descriptions into a workflow. In this lesson the differences between these two classes are explained,
how to pass data to and from command-line tools and specify working environments for these, and finally
how to use a tool description within a workflow.
You should follow the examples in this lesson from your novice-tutorial-exercises directory.
$ cd novice-tutorial-exercises
Our first CWL script
To demonstrate the basic requirements for a tool descriptor a CWL description for the popular “Hello world!” demonstration will be examined.
echo.cwl
cwlVersion: v1.2
class: CommandLineTool
baseCommand: echo
inputs:
message_text:
type: string
inputBinding:
position: 1
outputs: []
Next, the input file: hello_world.yml.
hello_world.yml
message_text: Hello world!
We will use the reference CWL runner, cwltool to run this CWL document (the .cwl workflow file) along with the .yml input file.
cwltool echo.cwl hello_world.yml
INFO Resolved 'echo.cwl' to 'file:///.../echo.cwl'
INFO [job echo.cwl] /private/tmp/docker_tmprm65mucw$ echo \
'Hello world!'
Hello world!
INFO [job echo.cwl] completed success
{}
INFO Final process status is success
The output displayed above shows that the program has run successfully and its output, Hello world!.
Let’s take a look at the echo.cwl script in more detail.
As explained above, the first 2 lines are always the same, the CWL version and the class of the script are defined.
In this example the class is CommandLineTool, in particular the echo command.
The next line, baseCommand, contains the command that will be run (echo).
inputs:
message_text:
type: string
inputBinding:
position: 1
This block of code contains the inputs section of the tool description. This section provides all the inputs that are needed for running this specific tool.
To run this example we will need to provide a string which will be included on the command line. Each of the inputs has a name, to help us tell them apart; this first input has the name : message_text.
The field inputBinding is one way to specify how the input should appear on the command line.
Here the position field indicates at which position the input will be on the command line; in this case the message_text value will be the first thing added to the command line (after the baseCommand, echo).
outputs: []
Lastly the outputs of the tool description. This example doesn’t have a formal output.
The text is printed directly in the terminal. So an empty YAML list ([]) is used as the output.
Script order
To make the script more readable the
inputfield is put in front of theoutputfield. However CWL syntax requires only that each field is properly defined, it does not require them to be in a particular order.
Changing input text
What do you need to change to print a different text on the command line?
Solution
To change the text on the command line, you only have to change the text in the
hello_world.ymlfile.For example:
message_text: Good job!
CWL single step workflow
The RNA-seq data from the introduction episode will be used for the first CWL workflow.
The first step of RNA-sequencing analysis is a quality control of the RNA reads using the fastqc tool.
This tool is already available to use so there is no need to write a new CWL tool description.
This is the workflow file (rna_seq_workflow_1.cwl).
rna_seq_workflow_1.cwl
cwlVersion: v1.2
class: Workflow
inputs:
rna_reads_fruitfly: File
steps:
quality_control:
run: bio-cwl-tools/fastqc/fastqc_2.cwl
in:
reads_file: rna_reads_fruitfly
out: [html_file]
outputs:
quality_report:
type: File
outputSource: quality_control/html_file
In a workflow the steps field must always be present. The workflow tasks or steps that you want to run are listed in this field.
At the moment the workflow only contains one step: quality_control. In the next episodes more steps will be added to the workflow.
Let’s take a closer look at the workflow. First the inputs field will be explained.
inputs:
rna_reads_fruitfly: File
Looking at the CWL script of the fastqc tool, it needs a fastq file as its input. In this example the fastq file consists of Drosophila melanogaster RNA reads.
So we call the variable rna_reads_fruitfly and it has File as its type.
To make this workflow interpretable for other researchers, self-explanatory and sensible variable names are used.
Input and output names
It is very important to give inputs and outputs a sensible name. Try not to use variable names like
inputAorinputBbecause others might not understand what is meant by it.
The next part of the script is the steps field.
steps:
quality_control:
run: bio-cwl-tools/fastqc/fastqc_2.cwl
in:
reads_file: rna_reads_fruitfly
out: [html_file]
Every step of a workflow needs a name, the first step of the workflow is called quality_control. Each step needs a run field, an in field and an out field.
The run field contains the location of the CWL file of the tool to be run. The in field connects the inputs field to the fastqc tool.
The fastqc tool has an input parameter called reads_file, so it needs to connect the reads_file to rna_reads_fruitfly.
Lastly, the out field is a list of output parameters from the tool to be used. In this example, the fastqc tool produces an output file called html_file.
The last part of the script is the output field.
outputs:
quality_report:
type: File
outputSource: quality_control/html_file
Each output in the outputs field needs its own name. In this example the output is called quality_report.
Inside quality_report the type of output is defined. The output of the quality_control step is a file, so the quality_report type is File.
The outputSource field refers to where the output is located, in this example it came from the step quality_control and it is called html_file.
When you want to run this workflow, you need to provide a file with the inputs the workflow needs. This file is similar to the hello_world.yml file in the previous section.
The input file is called workflow_input_1.yml
workflow_input_1.yml
rna_reads_fruitfly:
class: File
location: rnaseq/GSM461177_2_subsampled.fastqsanger
format: http://edamontology.org/format_1930 # FASTA
In the input file the values for the inputs that are declared in the inputs section of the workflow are provided.
The workflow takes rna_reads_fruitfly as an input parameter, so we use the same variable name in the input file.
When setting inputs, the class of the object needs to be defined, for example class: File or class: Directory.
The location field contains the location of the input file, in this case it is a local path, but
we could have directly used the original url location: https://zenodo.org/record/4541751/files/GSM461177_2_subsampled.fastqsanger
In this example the last line is needed to provide a format for the fastq file.
Now you can run the workflow using the following command:
cwltool --cachedir cache rna_seq_workflow_1.cwl workflow_input_1.yml
...
Analysis complete for GSM461177_2_subsampled.fastqsanger
INFO [job quality_control] Max memory used: 179MiB
INFO [job quality_control] completed success
INFO [step quality_control] completed success
INFO [workflow ] completed success
{
"quality_report": {
"location": "file:///.../GSM461177_2_subsampled.fastqsanger_fastqc.html",
"basename": "GSM461177_2_subsampled.fastqsanger_fastqc.html",
"class": "File",
"checksum": "sha1$e820c530b91a3087ae4c53a6f9fbd35ab069095c",
"size": 378324,
"path": "/.../GSM461177_2_subsampled.fastqsanger_fastqc.html"
}
}
INFO Final process status is success
Cache Directory
To save intermediate results for re-use later we use
--cachedir cache; wherecacheis the directory for storing the cache (it can be given any name, here we are just usingcachefor simplicity). You can safely delete thecachedirectory anytime, if you need to reclaim the disk space.
Exercise
Needs some exercises
- Ask the students to get some information from each report generated for the different data files we’ve downloaded. This will involve them making simple changes to the yaml configuration file.
Key Points
A tool description describes the interface to a command line tool.
A workflow describes which command line tools to use in one or more steps.
A tool descriptor is defined using the
ComandLineToolclass.FIXME: How can we use a tool descriptor in a single step workflow?
Developing Multi-Step Workflows
Overview
Teaching: 0 min
Exercises: 0 minQuestions
How can we expand to a multi-step workflow?
Iterative workflow development
Workflows as dependency graphs
How to use sketches for workflow design?
Objectives
explain that a workflow is a dependency graph
use cwlviewer online
generate Graphviz diagram using cwltool
exercise with the printout of a simple workflow; draw arrows on code; hand draw a graph on another sheet of paper
recognise that workflow development can be iterative i.e. that it doesn’t have to happen all at once
understand the flow of data between tools
Multi-Step Workflow
In the previous episode a single step workflow was shown, carrying out a simple RNA read
of the fruitfly genome. In this episode the workflow is extended with an equivalent reverse
RNA read and the next two steps of the RNA-sequencing analysis, trimming the reads and
aligning the trimmed reads, are added.
We will be using the cutadapt and STAR tools for these tasks.
To make a multi-step workflow that can carry all this analysis out, we add more entries
to the steps field. Note that when the quality_control step is duplicated the two
steps are named quality_control_forward and quality_control_reverse, to distinguish
the separate forward and reverse RNA reads. Likewise, the rna_reads_fruitfly input becomes
rna_reads_fruitfly_forward, and an rna_reads_fruitfly_reverse input is added.
rna_seq_workflow_2.cwl
cwlVersion: v1.2
class: Workflow
inputs:
rna_reads_fruitfly_forward:
type: File
format: http://edamontology.org/format_1930 # FASTQ
rna_reads_fruitfly_reverse:
type: File
format: http://edamontology.org/format_1930 # FASTQ
ref_fruitfly_genome: Directory
fruitfly_gene_model: File
steps:
quality_control_forward:
run: bio-cwl-tools/fastqc/fastqc_2.cwl
in:
reads_file: rna_reads_fruitfly_forward
out: [html_file]
quality_control_reverse:
run: bio-cwl-tools/fastqc/fastqc_2.cwl
in:
reads_file: rna_reads_fruitfly_reverse
out: [html_file]
trim_low_quality_bases:
run: bio-cwl-tools/cutadapt/cutadapt-paired.cwl
in:
reads_1: rna_reads_fruitfly_forward
reads_2: rna_reads_fruitfly_reverse
minimum_length: { default: 20 }
quality_cutoff: { default: 20 }
out: [ trimmed_reads_1, trimmed_reads_2, report ]
mapping_reads:
requirements:
ResourceRequirement:
ramMin: 5120
run: bio-cwl-tools/STAR/STAR-Align.cwl
in:
RunThreadN: {default: 4}
GenomeDir: ref_fruitfly_genome
ForwardReads: trim_low_quality_bases/trimmed_reads_1
ReverseReads: trim_low_quality_bases/trimmed_reads_2
OutSAMtype: {default: BAM}
SortedByCoordinate: {default: true}
OutSAMunmapped: {default: Within}
Overhang: { default: 36 } # the length of the reads - 1
Gtf: fruitfly_gene_model
out: [alignment]
index_alignment:
run: bio-cwl-tools/samtools/samtools_index.cwl
in:
bam_sorted: mapping_reads/alignment
out: [bam_sorted_indexed]
outputs:
quality_report_forward:
type: File
outputSource: quality_control_forward/html_file
quality_report_reverse:
type: File
outputSource: quality_control_reverse/html_file
bam_sorted_indexed:
type: File
outputSource: index_alignment/bam_sorted_indexed
The workflow file shows the first 5 steps of the RNA-seq analysis: quality_control_reverse,
quality_control_forward, trim_low_quality_bases, mapping_reads, and index_alignment.
The index_alignment step uses the alignment output of the mapping_reads step.
You do this by referencing the output of the mapping_reads step in the in field of the index_alignment step.
This is similar to referencing the outputs of the different steps in the outputs section.
The mapping_reads step needs some extra information beyond the inputs from the other steps, which
is done by providing default values. If you want, you can read the bio-cwl-tools/STAR/STAR-Align.cwl
file to see how these extra inputs are transformed into command line options to the STAR program.
This information is provided in the in field.
To run the tool better, it needs more RAM than the default. So there is a requirements entry inside
the mapping_reads step definition with a ResourceRequirement to allocate a minimum of
5120 MiB (5 GiB) of RAM.
The newly added mapping_reads step also need an input not provided by any of our other steps,
therefore an additional workflow-level input is added: a directory that contains the reference genome
necessary for the mapping.
This ref_fruitfly_genome is added in the inputs field of the workflow and in the YAML input file,
workflow_input_2.yml.
workflow_input_2.yml
rna_reads_fruitfly_forward:
class: File
location: rnaseq/GSM461177_1_subsampled.fastqsanger
format: http://edamontology.org/format_1930 # FASTQ
rna_reads_fruitfly_reverse:
class: File
location: rnaseq/GSM461177_2_subsampled.fastqsanger
format: http://edamontology.org/format_1930 # FASTQ
ref_fruitfly_genome:
class: Directory
location: rnaseq/dm6-STAR-index
fruitfly_gene_model:
class: File
location: rnaseq/Drosophila_melanogaster.BDGP6.87.gtf
Exercise
Draw the connecting arrows in the following graph of our workflow. Also, provide the outputs/inputs of the different steps. You can use for example Paint or print out the graph.
Solution
To find out how the inputs and the steps are connected to each other, you look at the
infield of the different steps.
Iterative working
Working on a workflow is often not something that happens all at once. Sometimes you already have a shell script ready that can be converted to a CWL workflow. Other times it is similar to this tutorial, you start with a single-step workflow and extend it to a multi-step workflow. This is all iterative working, a continuous work in progress.
Visualising a workflow
A CWL workflow is a directed acyclic graph (DAG). This means that:
- The workflow has a certain direction, from workflow inputs to step inputs, from step outputs to other step inputs, and from step outputs to workflow outputs and
- The workflow definition has no cycles.
A CWL workflow is a dependency graph. Each input for a step in the workflow depends on either a workflow-level input or a particular output from another step.
To visualise a workflow, a graph can be used. This can be done before a CWL script is written to
visualise how the different steps connect to each other. It is also possible to make a graph after
the CWL script has been written. This graph can be generated using online tools or the built-in
function in cwltool. When a graph is generated, it can be used to visualise the steps taken and
could make it easier to explain a workflow to other researchers.
From CWL script to graph
In this example the workflow is already made, so the graph can be generated using
cwlviewer online or using cwltool. First, let’s have a look at
cwlviewer. To use this tool, the workflow has to be put in a GitHub,
GitLab or Git repository. To view the graph of the workflow enter the URL and click Parse Workflow.
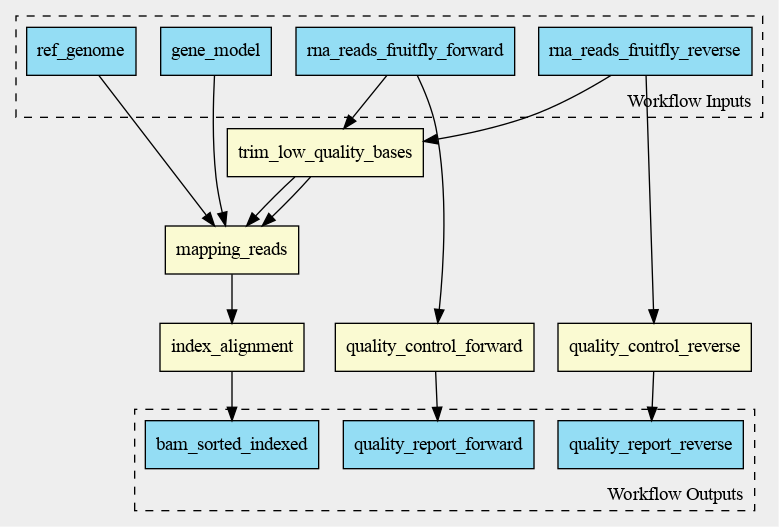
The cwlviewer displays the workflow as a graph, starting with the input. Then the different steps
are shown, each with their input(s) and output(s). The steps are linked to each other using arrows
accompanied by the input of the next step. The graph ends with the workflow outputs.
The graph of the RNA-seq workflow looks a follows:
It is also possible to generate the graph in the command line. cwltool has a function that makes a
graph. The --print-dot option will print a file suitable for Graphviz dot program. This is the
command to generate a Scalable Vector Graphic (SVG) file:
cwltool --print-dot rna_seq_workflow_2.cwl | dot -Tsvg > workflow_graph_2.svg
The resulting SVG file displays the same graph as the one in the cwlviewer. The SVG file can be opened in any web browser and in Inkscape, for example.
Visualisation in VSCode
Benten is an extension in Visual Studio Code (VSCode) that among other things visualises a workflow in a graph. When Benten is installed in VSCode, the tool can be used to visualise the workflow. In the top-right corner of the VSCode window the CWL viewer can be opened, see the screenshot below.
.png)
In VSCode/Benten the inputs are shown in green, the steps in blue and the outputs in yellow. This
graph looks a little bit different from the graph made with cwlviewer or cwltool.
The graph by VSCode/Benten doesn’t show the output-input names between the different steps.
.png)
Key Points
A multi-step workflow has multiple entries under the
stepssectionWorkflow development can be an iterative process
A CWL workflow can be represented as a dependency graph, either to explain your workflow or as a planning tool
Resources for Reusing Tools and Scripts
Overview
Teaching: 0 min
Exercises: 0 minQuestions
How to find other solutions/CWL recipes for awkward problems?
Objectives
Know good resources for finding solutions to common problems
Pre-written tool descriptions
When you start a CWL workflow, it is recommended to check if there is already a CWL document available for the tools you want to use. Bio-cwl-tools is a library of CWL documents for biology/life-sciences related tools.
The CWL documents of the previous steps were already provided for you, however, you can also find them in this library. In this episode you will use the bio-cwl-tools library to add the last step to the workflow.
Adding new step in workflow
The last step of our workflow is counting the RNA-seq reads for which we will use the featureCounts tool.
Exercise
Find the
featureCountstool in the bio-cwl-tools library. Have a look at the CWL document. Which inputs does this tool need? And what are the outputs of this tool?Solution
The
featureCountsCWL document can be found in the GitHub repo; it has 3 inputs:annotationsandmapped_reads, both files, along withreads_are_paired(a boolean choice). These inputs can be found on lines 6, 9, and 12. The output of this tool is a file calledfeaturecounts(line 27).
We need a local copy of featureCounts in order to use it in our workflow.
We already imported this as a git submodule during setup,
so the tool should be located at bio-cwl-tools/subread/featureCounts.cwl.
Exercise
Please copy the
rna_seq_workflow_2.cwlfile to createrna_seq_workflow_3.cwl. Add thefeatureCountstool to the workflow. Similar to theSTARtool, this tool also needs more RAM than the default. To run the tool a minimum of 500 MiB of RAM is needed. Use arequirementsentry withResourceRequirementto allocate aramMinof 500. Use the inputs and output of the previous exercise to connect this step to previous steps.Solution
rna_seq_workflow_3.cwl
cwlVersion: v1.2 class: Workflow inputs: rna_reads_fruitfly_forward: type: File format: http://edamontology.org/format_1930 # FASTQ rna_reads_fruitfly_reverse: type: File format: http://edamontology.org/format_1930 # FASTQ ref_fruitfly_genome: Directory fruitfly_gene_model: File steps: quality_control_forward: run: bio-cwl-tools/fastqc/fastqc_2.cwl in: reads_file: rna_reads_fruitfly_forward out: [html_file] quality_control_reverse: run: bio-cwl-tools/fastqc/fastqc_2.cwl in: reads_file: rna_reads_fruitfly_reverse out: [html_file] trim_low_quality_bases: run: bio-cwl-tools/cutadapt/cutadapt-paired.cwl in: reads_1: rna_reads_fruitfly_forward reads_2: rna_reads_fruitfly_reverse minimum_length: { default: 20 } quality_cutoff: { default: 20 } out: [ trimmed_reads_1, trimmed_reads_2, report ] mapping_reads: requirements: ResourceRequirement: ramMin: 5120 run: bio-cwl-tools/STAR/STAR-Align.cwl in: RunThreadN: {default: 4} GenomeDir: ref_fruitfly_genome ForwardReads: trim_low_quality_bases/trimmed_reads_1 ReverseReads: trim_low_quality_bases/trimmed_reads_2 OutSAMtype: {default: BAM} SortedByCoordinate: {default: true} OutSAMunmapped: {default: Within} Overhang: { default: 36 } # the length of the reads - 1 Gtf: fruitfly_gene_model out: [alignment] index_alignment: run: bio-cwl-tools/samtools/samtools_index.cwl in: bam_sorted: mapping_reads/alignment out: [bam_sorted_indexed] count_reads: requirements: ResourceRequirement: ramMin: 500 run: bio-cwl-tools/subread/featureCounts.cwl in: mapped_reads: index_alignment/bam_sorted_indexed annotations: fruitfly_gene_model reads_are_paired: {default: true} out: [featurecounts] outputs: quality_report_forward: type: File outputSource: quality_control_forward/html_file quality_report_reverse: type: File outputSource: quality_control_reverse/html_file bam_sorted_indexed: type: File outputSource: index_alignment/bam_sorted_indexed featurecounts: type: File outputSource: count_reads/featurecounts
The workflow is complete and we only need to complete the YAML input file.
Please copy the workflow_input_2.yml file to workflow_input_3.yml, and
add the last entry in the input file, which is the fruitfly_gene_model file.
workflow_input_3.yml
rna_reads_fruitfly_forward:
class: File
location: rnaseq/GSM461177_1_subsampled.fastqsanger
format: http://edamontology.org/format_1930 # FASTQ
rna_reads_fruitfly_reverse:
class: File
location: rnaseq/GSM461177_2_subsampled.fastqsanger
format: http://edamontology.org/format_1930 # FASTQ
ref_fruitfly_genome:
class: Directory
location: rnaseq/dm6-STAR-index
fruitfly_gene_model:
class: File
location: rnaseq/Drosophila_melanogaster.BDGP6.87.gtf
format: http://edamontology.org/format_2306
You have finished the workflow and the input file and now you can run the whole workflow.
cwltool --cachedir cache rna_seq_workflow_3.cwl workflow_input_3.yml
Key Points
bio-cwl-tools is a library of CWL documents for biology/life-sciences related tools
Debugging Workflows
Overview
Teaching: 0 min
Exercises: 0 minQuestions
How can I check my CWL file for errors?
How can I get more information to help with solving an error?
What are some common error messages when using CWL?
Objectives
Check a CWL file for errors
Output debugging information
Interpret and fix commonly encountered error messages
When working on a CWL workflow, you will probably encounter errors. There are many different errors possible. It is always very important to check the error message in the terminal, because it will give you information on the error. This error message will give you the type of error as well as the line of code that contains the error. Some of these errors will be explained in this episode.
As a first step to check if your CWL script contains any errors, you can run the workflow with the --validate flag.
cwltool --validate CWL_SCRIPT.cwl
It is possible that the script is validated, however, it still gets an error.
If you encounter an error, the best practice is to run the workflow with the --debug flag.
This will provide you with extensive information on the error you encounter.
cwltool --debug CWL_SCRIPT.cwl
YAML errors
First of all, errors in the YAML syntax. When writing a piece of code, it is very easy to make a mistake.
Some very common YAML errors are:
Tabs
Using tabs instead of spaces. In YAML files indentations are made using spaces, not tabs. Please download and run this example which includes a tab character.
$ cwltool tab-error.cwl workflow_input.yml
ERROR Tool definition failed validation:
while scanning for the next token
file:///tab-error.cwl:5:1: found character '\t' that cannot start any token
Field Name Typos
Typos in field names. It is very easy to forget for example the capital letters in field names.
Errors with typos in field names will show invalid field.
rna_seq_workflow_fieldname_fail.cwl
cwlVersion: v1.2
class: Workflow
inputs:
rna_reads_fruitfly: File
ref_fruitfly_genome: Directory
steps:
quality_control:
run: bio-cwl-tools/fastqc/fastqc_2.cwl
in:
reads_file: rna_reads_fruitfly
out: [html_file]
mapping_reads:
requirements:
ResourceRequirement:
ramMin: 5120
run: bio-cwl-tools/STAR/STAR-Align.cwl
in:
RunThreadN: {default: 4}
GenomeDir: ref_fruitfly_genome
ForwardReads: rna_reads_fruitfly
OutSAMtype: {default: BAM}
SortedByCoordinate: {default: true}
OutSAMunmapped: {default: Within}
out: [alignment]
index_alignment:
run: bio-cwl-tools/samtools/samtools_index.cwl
in:
bam_sorted: mapping_reads/alignment
out: [bam_sorted_indexed]
outputs:
qc_html:
type: File
outputsource: quality_control/html_file
bam_sorted_indexed:
type: File
outputSource: index_alignment/bam_sorted_indexed
workflow_input_debug.yml
rna_reads_fruitfly:
class: File
location: rnaseq/GSM461177_1_subsampled.fastqsanger
format: http://edamontology.org/format_1930 # FASTQ
ref_fruitfly_genome:
class: Directory
location: rnaseq/dm6-STAR-index
$ cwltool rna_seq_workflow_fieldname_fail.cwl workflow_input_debug.yml
ERROR Tool definition failed validation:
rna_seq_workflow_fieldname_fail.cwl:1:1: Object `rna_seq_workflow_fieldname_fail.cwl` is not valid
because
tried `Workflow` but
rna_seq_workflow_fieldname_fail.cwl:35:1: the `outputs` field is not valid because
rna_seq_workflow_fieldname_fail.cwl:36:3: item is invalid because
rna_seq_workflow_fieldname_fail.cwl:38:5: invalid field `outputsource`, expected one of:
'label', 'secondaryFiles', 'streamable', 'doc', 'id',
'format', 'outputSource', 'linkMerge', 'pickValue', 'type'
Variable Name Typos
Typos in variable names. Similar to typos in field names, it is easy to make a mistake in referencing to a variable.
These errors will show Field references unknown identifier.
rna_seq_workflow_varname_fail.cwl
cwlVersion: v1.2
class: Workflow
inputs:
rna_reads_fruitfly: File
ref_fruitfly_genome: Directory
steps:
quality_control:
run: bio-cwl-tools/fastqc/fastqc_2.cwl
in:
reads_file: rna_reads_fruitfly
out: [html_file]
mapping_reads:
requirements:
ResourceRequirement:
ramMin: 5120
run: bio-cwl-tools/STAR/STAR-Align.cwl
in:
RunThreadN: {default: 4}
GenomeDir: ref_fruitfly_genome
ForwardReads: rna_reads_fruitfly
OutSAMtype: {default: BAM}
SortedByCoordinate: {default: true}
OutSAMunmapped: {default: Within}
out: [alignment]
index_alignment:
run: bio-cwl-tools/samtools/samtools_index.cwl
in:
bam_sorted: mapping_reads/alignments
out: [bam_sorted_indexed]
outputs:
qc_html:
type: File
outputSource: quality_control/html_file
bam_sorted_indexed:
type: File
outputSource: index_alignment/bam_sorted_indexed
$ cwltool rna_seq_workflow_varname_fail.cwl workflow_input_debug.yml
ERROR Tool definition failed validation:
rna_seq_workflow_varname_fail.cwl:8:1: checking field `steps`
rna_seq_workflow_varname_fail.cwl:29:3: checking object
`rna_seq_workflow_varname_fail.cwl#index_alignment`
rna_seq_workflow_varname_fail.cwl:31:5: checking field `in`
rna_seq_workflow_varname_fail.cwl:32:7: checking object
`rna_seq_workflow_varname_fail.cwl#index_alignment/bam_sorted`
Field `source` references unknown identifier
`mapping_reads/alignments`, tried
file:///.../rna_seq_workflow_varname_fail.cwl#mapping_reads/alignments
Wiring error
Wiring errors often occur when you forget to add an output from a workflow’s step to the outputs section.
This doesn’t cause an error message, but there won’t be any output in your directory.
To get the desired output you have to run the workflow again.
Best practice is to check your outputs section before running your script to make sure all the outputs you want are there.
Type mismatch
Type errors take place when there is a mismatch in type between variables.
When you declare a variable in the inputs section, the type of this variable has to match the type in the YAML inputs file
and the type used in one of the workflows steps.
The error message that is shown when this error occurs will tell you on which line the mismatch happens.
rna_seq_workflow_type_fail.cwl
cwlVersion: v1.2
class: Workflow
inputs:
rna_reads_fruitfly: int
ref_fruitfly_genome: Directory
steps:
quality_control:
run: bio-cwl-tools/fastqc/fastqc_2.cwl
in:
reads_file: rna_reads_fruitfly
out: [html_file]
mapping_reads:
requirements:
ResourceRequirement:
ramMin: 5120
run: bio-cwl-tools/STAR/STAR-Align.cwl
in:
RunThreadN: {default: 4}
GenomeDir: ref_fruitfly_genome
ForwardReads: rna_reads_fruitfly
OutSAMtype: {default: BAM}
SortedByCoordinate: {default: true}
OutSAMunmapped: {default: Within}
out: [alignment]
index_alignment:
run: bio-cwl-tools/samtools/samtools_index.cwl
in:
bam_sorted: mapping_reads/alignment
out: [bam_sorted_indexed]
outputs:
qc_html:
type: File
outputSource: quality_control/html_file
bam_sorted_indexed:
type: File
outputSource: index_alignment/bam_sorted_indexed
$ cwltool rna_seq_workflow_type_fail.cwl workflow_input_debug.yml
ERROR Tool definition failed validation:
rna_seq_workflow_type_fail.cwl:5:3: Source 'rna_reads_fruitfly' of type "int" is incompatible
rna_seq_workflow_type_fail.cwl:12:7: with sink 'reads_file' of type "File"
rna_seq_workflow_type_fail.cwl:5:3: Source 'rna_reads_fruitfly' of type "int" is incompatible
rna_seq_workflow_type_fail.cwl:23:7: with sink 'ForwardReads' of type ["File", {"type":
"array", "items": "File"}]
Format error
Some files need a specific format that needs to be specified in the YAML inputs file, for example the fastq file in the RNA-seq analysis. When you don’t specify a format, an error will occur. You can for example use the EDAM ontology.
rna_seq_workflow_debug.cwl
cwlVersion: v1.2
class: Workflow
inputs:
rna_reads_fruitfly: File
ref_fruitfly_genome: Directory
steps:
quality_control:
run: bio-cwl-tools/fastqc/fastqc_2.cwl
in:
reads_file: rna_reads_fruitfly
out: [html_file]
mapping_reads:
requirements:
ResourceRequirement:
ramMin: 5120
run: bio-cwl-tools/STAR/STAR-Align.cwl
in:
RunThreadN: {default: 4}
GenomeDir: ref_fruitfly_genome
ForwardReads: rna_reads_fruitfly
OutSAMtype: {default: BAM}
SortedByCoordinate: {default: true}
OutSAMunmapped: {default: Within}
out: [alignment]
index_alignment:
run: bio-cwl-tools/samtools/samtools_index.cwl
in:
bam_sorted: mapping_reads/alignment
out: [bam_sorted_indexed]
outputs:
qc_html:
type: File
outputSource: quality_control/html_file
bam_sorted_indexed:
type: File
outputSource: index_alignment/bam_sorted_indexed
workflow_input_undefined.yml
rna_reads_fruitfly:
class: File
location: rnaseq/GSM461177_1_subsampled.fastqsanger
ref_fruitfly_genome:
class: Directory
location: rnaseq/dm6-STAR-index
$ cwltool rna_seq_workflow_debug.cwl workflow_input_undefined.yml
ERROR Exception on step 'mapping_reads'
ERROR [step mapping_reads] Cannot make job: Expected value of 'ForwardReads' to have format http://edamontology.org/format_1930 but
File has no 'format' defined: {
"class": "File",
"location": "file:///.../rnaseq/GSM461177_1_subsampled.fastqsanger",
"size": 142867948,
"basename": "GSM461177_1_subsampled.fastqsanger",
"nameroot": "GSM461177_1_subsampled",
"nameext": ".fastqsanger"
}
Key Points
Run the workflow with the
--validateoption to check for errorsThe
--debugoption will output more information‘Wiring’ errors won’t necessarily yield an error message
More information
Overview
Teaching: min
Exercises: minQuestions
Objectives
If you want to know more about CWL script and workflows, you can look at one of these websites:
- CWL User Guide
- YAML Guide
- Extra CWL Command Line Tool information
- Miscellaneous CWL information
- Recommended Practices in CWL
Key Points