Content from Running commands with Maestro
Last updated on 2024-08-22 | Edit this page
Estimated time: 60 minutes
Overview
Questions
- “How do I run a simple command with Maestro?”
Objectives
- “Create a Maestro YAML file”
What is the workflow I’m interested in?
In this lesson we will make an experiment that takes an application which runs in parallel and investigate it’s scalability. To do that we will need to gather data, in this case that means running the application multiple times with different numbers of CPU cores and recording the execution time. Once we’ve done that we need to create a visualization of the data to see how it compares against the ideal case.
From the visualization we can then decide at what scale it makes most sense to run the application at in production to maximize the use of our CPU allocation on the system.
We could do all of this manually, but there are useful tools to help us manage data analysis pipelines like we have in our experiment. Today we’ll learn about one of those: Maestro.
In order to get started with Maestro, let’s begin by taking a simple
command and see how we can run that via Maestro. Let’s choose the
command hostname which prints out the name of the host
where the command is executed:
OUTPUT
pascal83That prints out the result but Maestro relies on files to know the status of your workflow, so let’s redirect the output to a file:
Writing a Maestro YAML
Edit a new text file named hostname.yaml. The file
extension is a recursive initialism for “YAML
Ain’t Markup Language”, a popular format for configuration files and
key-value data serialization. For more, see the Wikipedia page, esp. YAML Syntax.
Contents of hostname.yaml (spaces matter!):
YML
description:
name: Hostnames
description: Report a node's hostname.
study:
- name: hostname-login
description: Write the login node's hostname to a file.
run:
cmd: |
hostname > hostname_login.txtKey points about this file
- The name of
hostname.yamlis not very important; it gives us information about file contents and type, but maestro will behave the same if you rename it tohostnameorfoo.txt. - The file specifies fields in a hierarchy. For example,
name,description, andrunare all passed tostudyand are at the same level in the hierarchy.descriptionandstudyare both at the top level in the hierarchy. - Indentation indicates the hierarchy and should be consistent. For
example, all the fields passed directly to
studyare indented relative tostudyand their indentation is all the same. - The commands executed during the study are given under
cmd. Starting this entry with|and a newline character allows us to specify multiple commands. - The example YAML file above is pretty minimal; all fields shown are required.
- The names given to
studycan include letters, numbers, and special characters.
Back in the shell we’ll run our new rule. At this point, we may see an error if a required field is missing or if our indentation is inconsistent.
bash: maestro: command not found...
If your shell tells you that it cannot find the command
maestro then we need to make the software available
somehow. In our case, this means activating the python virtual
environment where maestro is installed.
You can tell this command has already been run when
(maestro_venv) appears before your command prompt:
BASH
janeh@pascal83:~$ source /usr/global/docs/training/janeh/maestro_venv/bin/activate
(maestro_venv) janeh@pascal83:~$Now that the maestro_venv virtual environment has been
activated, the maestro command should be available, but
let’s double check
OUTPUT
/usr/global/docs/training/janeh/maestro_venv/bin/maestroRunning maestro
Once you have maestro available to you, run
maestro run hostname.yaml and enter y when
prompted:
OUTPUT
[2024-03-20 15:39:34: INFO] INFO Logging Level -- Enabled
[2024-03-20 15:39:34: WARNING] WARNING Logging Level -- Enabled
[2024-03-20 15:39:34: CRITICAL] CRITICAL Logging Level -- Enabled
[2024-03-20 15:39:34: INFO] Loading specification -- path = hostname.yaml
[2024-03-20 15:39:34: INFO] Directory does not exist. Creating directories to ~/Hostnames_20240320-153934/logs
[2024-03-20 15:39:34: INFO] Adding step 'hostname-login' to study 'Hostnames'...
[2024-03-20 15:39:34: INFO]
------------------------------------------
Submission attempts = 1
Submission restart limit = 1
Submission throttle limit = 0
Use temporary directory = False
Hash workspaces = False
Dry run enabled = False
Output path = ~/Hostnames_20240320-153934
------------------------------------------
Would you like to launch the study? [yn] y
Study launched successfully.and look at the outputs. You should have a new directory whose name
includes a date and timestamp and that starts with the name
given under description at the top of
hostname.yaml.
In that directory will be a subdirectory for every study
run from hostname.yaml. The subdirectories for each study
include all output files for that study.
BASH
(maestro_venv) janeh@pascal83:~$ cd Hostnames_20240320-153934/
(maestro_venv) janeh@pascal83:~/Hostnames_20240320-153934$ lsOUTPUT
batch.info Hostnames.pkl Hostnames.txt logs status.csv
hostname-login Hostnames.study.pkl hostname.yaml metaBASH
(maestro_venv) janeh@pascal83:~/Hostnames_20240320-153934$ cd hostname-login/
(maestro_venv) janeh@pascal83:~/Hostnames_20240320-153934/hostname-login$ lsOUTPUT
hostname-login.2284862.err hostname-login.2284862.out hostname-login.sh hostname_login.txtChallenge
To which file will the login node’s hostname, pascal83,
be written?
hostname-login.2284862.errhostname-login.2284862.outhostname-login.shhostname_login.txt
- hostname_login.txt
In the original hostname.yaml file that we ran, we
specified that hostname would be written to
hostname_login.txt, and this is where we’ll see that
output, if the run worked!
Challenge
This one is tricky! In the example above, pascal83 was
written to
~/Hostnames_{date}_{time}/hostname-login/hostname_login.txt.
Where would Hello be written for the following YAML?
YML
description:
name: MyHello
description: Report a node's hostname.
study:
- name: give-salutation
description: Write the login node's hostname to a file
run:
cmd: |
echo "hello" > greeting.txt~/give-salutation_{date}_{time}/greeting/greeting.txt~/greeting_{date}_{time}/give_salutation/greeting.txt~/MyHello_{date}_{time}/give-salutation/greeting.txt~/MyHello_{date}_{time}/greeting/greeting.txt
.../MyHello_{date}_{time}/give-salutation/greeting.txt
The top-level folder created starts with the name field
under description; here, that’s MyHello. Its
subdirectory is named after the study; here, that’s
give-salutation. The file created is
greeting.txt and this stores the output of
echo "hello".
Callout
After running a workflow with Maestro, you can check the status via
maestro status --disable-theme <directory name>. For
example, for the directory Hostnames_20240821-165341
created via maestro run hostnames.yaml:
OUTPUT
Study: /usr/WS1/janeh/maestro-tut/Hostnames_20240821-165341
┏━━━━━━━━━━━━┳━━━━━━━━━┳━━━━━━━━━━━┳━━━━━━━━━━┳━━━━━━━━━━━━┳━━━━━━━━━━━┳━━━━━━━━━━━━┳━━━━━━━━━━━┳━━━━━━━━━━━━┳━━━━━━━━━━━┓
┃ ┃ ┃ ┃ ┃ ┃ Elapsed ┃ ┃ Submit ┃ ┃ Number ┃
┃ Step Name ┃ Job ID ┃ Workspace ┃ State ┃ Run Time ┃ Time ┃ Start Time ┃ Time ┃ End Time ┃ Restarts ┃
┡━━━━━━━━━━━━╇━━━━━━━━━╇━━━━━━━━━━━╇━━━━━━━━━━╇━━━━━━━━━━━━╇━━━━━━━━━━━╇━━━━━━━━━━━━╇━━━━━━━━━━━╇━━━━━━━━━━━━╇━━━━━━━━━━━┩
│ hostname-l │ 2593210 │ hostname- │ FINISHED │ 0d:00h:00m │ 0d:00h:00 │ 2024-08-21 │ 2024-08-2 │ 2024-08-21 │ 0 │
│ ogin │ │ login │ │ :01s │ m:01s │ 16:53:44 │ 1 │ 16:53:45 │ │
│ │ │ │ │ │ │ │ 16:53:44 │ │ │
└────────────┴─────────┴───────────┴──────────┴────────────┴───────────┴────────────┴───────────┴────────────┴───────────┘
(END)Key Points
- You execute
maestro runwith a YAML file including information about your run. - Your run includes a description and at least one study (a step in your run).
- Your maestro run creates a directory with subdirectories and outputs for each study.
- Check the status of a run via
maestro status --disable-theme <directory>
Content from Running Maestro on the cluster
Last updated on 2024-06-06 | Edit this page
Estimated time: 50 minutes
How do I run Maestro on the cluster?
What happens when we want to run on the cluster (“to run a batch job”) rather than the login node? The cluster we are using uses Slurm, and Maestro has built in support for Slurm. We just need to tell Maestro which resources we need Slurm to grab for our run.
First, we need to add a batch block to our YAML file,
where we’ll provide the names of the machine, bank, and queue in which
your jobs should run.
YML
batch:
type: slurm
host: ruby # enter the machine you'll run on
bank: guests # enter the bank to charge
queue: pbatch # partition in which your job should run
reservation: HPCC1B # reservation for this workshopSecond, we need to specify the number of nodes, number of processes,
and walltime for each step in our YAML file. This information
goes under each step’s run field. Here we specify 1 node, 1
process, and a time limit of 30 seconds:
Whereas run previously held only info about the command
we wanted to execute, steps run on the cluster include a specification
of the resources needed in order to execute. Note that
the format of the walltime includes quotation marks –
"{Hours}:{Minutes}:{Seconds}".
With these changes, our updated YAML file might look like
YML
---
description:
name: Hostnames
description: Report a node's hostname.
batch:
type: slurm
host: ruby # machine
bank: guests # bank
queue: pbatch # partition
reservation: HPCC1B # reservation for this workshop
study:
- name: hostname-login
description: Write the login node's hostname to a file
run:
cmd: |
hostname > hostname_login.txt
- name: hostname_batch
description: Write the node's hostname to a file
run:
cmd: |
hostname >> hostname.txt
nodes: 1
procs: 1
walltime: "00:00:30"Note that we left the rule hostname-login as is. Because
we do not specify any info for slurm under this original step’s
run field – like nodes, processes, or walltime – this step
will continue running on the login node and only
hostname_batch will be handed off to slurm.
Running on the cluster
Modify your YAML file, hostname.yaml to execute
hostname on the cluster. Run with 1 node and 1
process using the bank guests on the partition
pbatch on ruby.
If you run this multiple times, do you always run on the same node? (Is the hostname printed always the same?)
The contents of hostname.yaml should look something
like:
YML
description:
name: Hostnames
description: Report a node's hostname.
batch:
type: slurm
host: ruby # machine
bank: guests # bank
queue: pbatch # partition
reservation: HPCC1B # reservation for this workshop
study:
- name: hostname-login
description: Write the login node's hostname to a file
run:
cmd: |
hostname > hostname_login.txt
- name: hostname_batch
description: Write the node's hostname to a file
run:
cmd: |
hostname >> hostname.txt
nodes: 1
procs: 1
walltime: "00:00:30"Go ahead and run the job:
A directory called Hostname_... will be created. If you
look in the subdirectory hostname_batch, you’ll find a file
called hostname.txt with info about the compute node where
the hostname command ran. If you run the job multiple
times, you will probably land on different nodes; this means you’ll see
different node numbers in different hostname.txt files. If
you see the same number more than once, don’t worry! (If you want to
double check that the hostnames printed are not for login nodes, you can
run nodeattr -c login to check the IDs of all login nodes
on the system.)
Outputs from a batch job
When running in batch, maestro run ... will create a new
directory with the same naming scheme as seen in episode 1, and that
directory will contain subdirectories for all studies. The
hostname_batch subdirectory has four output files, but this
time the file ending with extension .sh is a slurm
submission script
OUTPUT
hostname.err hostname.out hostname.slurm.sh hostname.txtBASH
#!/bin/bash
#SBATCH --nodes=1
#SBATCH --partition=pvis
#SBATCH --account=lc
#SBATCH --time=00:00:30
#SBATCH --job-name="hostname"
#SBATCH --output="hostname.out"
#SBATCH --error="hostname.err"
#SBATCH --comment "Write the node's hostname to a file"
hostname > hostname.txtMaestro uses the info from your YAML file to write this script and
then submits it to the scheduler for you. Soon after you run on the
cluster via maestro run hostname.yaml, you should be able
to see the job running or finishing up in the queue with the command
squeue -u «your username».
OUTPUT
[2024-03-20 17:31:37: INFO] INFO Logging Level -- Enabled
[2024-03-20 17:31:37: WARNING] WARNING Logging Level -- Enabled
[2024-03-20 17:31:37: CRITICAL] CRITICAL Logging Level -- Enabled
[2024-03-20 17:31:37: INFO] Loading specification -- path = batch-hostname.yaml
[2024-03-20 17:31:37: INFO] Directory does not exist. Creating directories to ~/Hostnames_20240320-173137/logs
[2024-03-20 17:31:37: INFO] Adding step 'hostname-login' to study 'Hostnames'...
[2024-03-20 17:31:37: INFO] Adding step 'hostname_batch' to study 'Hostnames'...
[2024-03-20 17:31:37: INFO]
------------------------------------------
Submission attempts = 1
Submission restart limit = 1
Submission throttle limit = 0
Use temporary directory = False
Hash workspaces = False
Dry run enabled = False
Output path = ~/Hostnames_20240320-173137
------------------------------------------
Would you like to launch the study? [yn] y
Study launched successfully.OUTPUT
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
718308 pvis hostname janeh R 0:01 1 pascal13Key Points
- You can run on the cluster by including info for Slurm in your Maestro YAML file
- Maestro generates and submits its own batch scripts to your scheduler.
- Steps without Slurm parameters will run on the login node by default.
Content from MPI applications and Maestro
Last updated on 2024-06-04 | Edit this page
Estimated time: 50 minutes
Overview
Questions
- “How do I run an MPI application via Maestro on the cluster?”
Objectives
- “Define rules to run parallel applications on the cluster”
Now it’s time to start getting back to our real workflow. We can execute a command on the cluster, but how do we effectively leverage cluster resources and actually run in parallel? In this episode, we’ll learn how to execute an application that can be run in parallel.
Our application is called amdahl and is available in the
python virtual environment we’re already using for maestro.
Check that you have access to this binary by running
which amdahl at the command line. You should see something
like
OUTPUT
/usr/global/docs/training/janeh/maestro_venv/bin/amdahlWe’ll use this binary to see how efficiently we can run a parallel program on our cluster – i.e. how the amount of work done per processor changes as we use more processors.
You should see output that looks roughly like
OUTPUT
Doing 30.000000 seconds of 'work' on 1 processor,
which should take 30.000000 seconds with 0.800000
parallel proportion of the workload.
Hello, World! I am process 0 of 1 on pascal83.
I will do all the serial 'work' for 5.243022 seconds.
Hello, World! I am process 0 of 1 on pascal83.
I will do parallel 'work' for 25.233023 seconds.
Total execution time (according to rank 0): 30.537750 secondsIn short, this program prints the amount of time spent working serially and the amount of time it spends on the parallel section of a code. On the login node, only a single task is created, so there shouldn’t be any speedup from running in parallel, but soon we’ll use more tasks to run this program!
In the last challenge, we saw how the amdahl executable
behaves when run on the login node. In the next challenge, let’s get
amdahl running on the login node using
maestro.
Challenge
Using what you learned in episode 1, create a Maestro YAML file that
runs amdahl on the login node and captures the output in a
file.
Next, let’s get amdahl running in batch, on a compute
node.
Challenge
Update amdahl.yaml from the last challenge so that this
workflow runs on a compute node with a single task. Use the examples
from episode 2!
Once you’ve done this. Examine the output and verify that only a single task is reporting on its work in your output file.
The contents of amdahl.yaml should now look something
like
YML
description:
name: Amdahl
description: Run on the cluster
batch:
type: slurm
host: ruby # machine
bank: guests # bank
queue: pbatch # partition
reservation: HPCC1B # reservation for this workshop
study:
- name: amdahl
description: run on the cluster
run:
cmd: |
amdahl >> amdahl.out
nodes: 1
procs: 1
walltime: "00:00:30"In your amdahl.out file, you should see that only a
single task – task 0 of 1 – is mentioned.
Note – Exact wording for names and descriptions is not important, but should help you to remember what this file and its study are doing.
Challenge
After checking that amdahl.yaml looks similar to the
solution above, update the number of nodes and
procs each to 2 and rerun
maestro run amdahl.yaml. How many processes report their
work in the output file?
Your YAML files contents are now updated to
YML
description:
name: Amdahl
description: Run on the cluster
batch:
type: slurm
host: ruby # machine
bank: guests # bank
queue: pbatch # partition
reservation: HPCC1B # reservation for this workshop
study:
- name: amdahl
description: run on the cluster
run:
cmd: |
amdahl >> amdahl.out
nodes: 2
procs: 2
walltime: "00:00:30"In the study folder, amdahl.slurm.sh will look something
like
BASH
#!/bin/bash
#SBATCH --nodes=2
#SBATCH --partition=pdebug
#SBATCH --account=guests
#SBATCH --time=00:01:00
#SBATCH --job-name="amdahl"
#SBATCH --output="amdahl.out"
#SBATCH --error="amdahl.err"
#SBATCH --comment "run Amdahl on the cluster"
amdahl >> amdahl.outand in amdahl.out, you probably see something like
OUTPUT
Doing 30.000000 seconds of 'work' on 1 processor,
which should take 30.000000 seconds with 0.800000
parallel proportion of the workload.
Hello, World! I am process 0 of 1 on pascal17.
I will do all the serial 'work' for 5.324555 seconds.
Hello, World! I am process 0 of 1 on pascal17.
I will do parallel 'work' for 22.349517 seconds.
Total execution time (according to rank 0): 27.755552 secondsNotice that this output refers to only “1 processor” and mentions
only one process. We requested two processes, but only a single one
reports back! Additionally, we requested two nodes, but only
one is mentioned in the above output (pascal17).
So what’s going on?
If your job were really using both tasks and nodes that were
assigned to it, then both would have written to
amdahl.out.
The amdahl binary is enabled to run in parallel but it’s
also able to run in serial. If we want it to run in parallel, we’ll have
to tell it so more directly.
Here’s the takeaway from the challenges above: It’s not enough to have both parallel resources and a binary/executable/program that is enabled to run in parallel. We actually need to invoke MPI in order to force our parallel program to use parallel resources.
Maestro and MPI
We didn’t really run an MPI application in the last section as we only ran on one processor. How do we request to run using multiple processes for a single step?
The answer is that we have to tell Slurm that we want to use MPI. In
the Intro to HPC lesson, the episodes introducing Slurm and running
parallel jobs showed that commands to run in parallel need to use
srun. srun talks to MPI and allows multiple
processors to coordinate work. A call to srun might look
something like
To make this easier, Maestro offers the shorthand
$(LAUNCHER). Maestro will replace instances of
$(LAUNCHER) with a call to srun, specifying as
many nodes and processes we’ve already told Slurm we want to use.
Challenge
Update amdahl.yaml to include $(LAUNCHER)
in the call to amdahl so that your study’s cmd
field includes
Run maestro with the updated YAML and explore the outputs. How many
tasks are mentioned in amdahl.out? In the Slurm submission
script created by Maestro (included in the same subdirectory as
amdahl.out), what text was used to replace
$(LAUNCHER)?
The updated YAML should look something like
YML
description:
name: Amdahl
description: Run a parallel program
batch:
type: slurm
host: ruby # machine
bank: guests # bank
queue: pbatch # partition
reservation: HPCC1B # reservation for this workshop
study:
- name: amdahl
description: run in parallel
run:
# Here's where we include our MPI wrapper:
cmd: |
$(LAUNCHER) amdahl >> amdahl.out
nodes: 2
procs: 2
walltime: "00:00:30"Your output file Amdahl_.../amdahl/amdahl.out should
include “Doing 30.000000 seconds of ‘work’ on 2 processors” and the
submission script Amdahl_.../amdahl/amdahl.slurm.sh should
include the line srun -n 2 -N 2 amdahl >> amdahl.out.
Maestro substituted srun -n 2 -N 2 for
$(LAUNCHER)!
Commenting Maestro YAML files
In the solution from the last challenge, the line beginning
# is a comment line. Hopefully you are already in the habit
of adding comments to your own scripts. Good comments make any script
more readable, and this is just as true with our YAML files.
Customizing amdahl output
Another thing about our application amdahl is that we
ultimately want to process the output to generate our scaling plot. The
output right now is useful for reading but makes processing harder.
amdahl has an option that actually makes this easier for
us. To see the amdahl options we can use
OUTPUT
usage: amdahl [-h] [-p [PARALLEL_PROPORTION]] [-w [WORK_SECONDS]] [-t] [-e]
options:
-h, --help show this help message and exit
-p [PARALLEL_PROPORTION], --parallel-proportion [PARALLEL_PROPORTION]
Parallel proportion should be a float between 0 and 1
-w [WORK_SECONDS], --work-seconds [WORK_SECONDS]
Total seconds of workload, should be an integer > 0
-t, --terse Enable terse output
-e, --exact Disable random jitterThe option we are looking for is --terse, and that will
make amdahl print output in a format that is much easier to
process, JSON. JSON format in a file typically uses the file extension
.json so let’s add that option to our shell
command and change the file format of the output
to match our new command:
YML
description:
name: Amdahl
description: Run a parallel program
batch:
type: slurm
host: ruby # machine
bank: guests # bank
queue: pbatch # partition
reservation: HPCC1B # reservation for this workshop
study:
- name: amdahl
description: run in parallel
run:
# Here's where we include our MPI wrapper:
cmd: |
$(LAUNCHER) amdahl --terse >> amdahl.json
nodes: 2
procs: 2
walltime: "00:01:30"There was another parameter for amdahl that caught my
eye. amdahl has an option
--parallel-proportion (or -p) which we might
be interested in changing as it changes the behavior of the code, and
therefore has an impact on the values we get in our results. Let’s try
specifying a parallel proportion of 90%:
YML
description:
name: Amdahl
description: Run a parallel program
batch:
type: slurm
host: ruby # machine
bank: guests # bank
queue: pbatch # partition
reservation: HPCC1B # reservation for this workshop
study:
- name: amdahl
description: run in parallel
run:
# Here's where we include our MPI wrapper:
cmd: |
$(LAUNCHER) amdahl --terse -p .9 >> amdahl.json
nodes: 2
procs: 2
walltime: "00:00:30"Challenge
Create a YAML file for a value of -p of 0.999 (the
default value is 0.8) for the case where we have a single node and 4
parallel processes. Run this workflow with Maestro to make sure your
script is working.
YML
description:
name: Amdahl
description: Run a parallel program
batch:
type: slurm
host: ruby # machine
bank: guests # bank
queue: pbatch # partition
reservation: HPCC1B # reservation for this workshop
study:
- name: amdahl
description: run in parallel
run:
# Here's where we include our MPI wrapper:
cmd: |
$(LAUNCHER) amdahl --terse -p .999 >> amdahl.json
nodes: 1
procs: 4
walltime: "00:00:30"Environment variables
Our current directory is probably starting to fill up with
directories starting with Amdahl_..., distinguished only by
dates and timestamps. It’s probably best to group runs into separate
folders to keep things tidy. One way we can do this is by specifying an
env section in our YAML file with a variable called
OUTPUT_PATH specified in this format:
This env block goes above our study block;
env is at the same level of indentation as
study. In this case, directories created by runs using this
OUTPUT_PATH will all be grouped inside the directory
Episode3, to help us group runs by where we are in the
lesson.
Challenge
Modify your YAML so that subsequent runs will be grouped into a
shared parent directory (for example, Episode3, as
above).
YML
description:
name: Amdahl
description: Run a parallel program
batch:
type: slurm
host: ruby # machine
bank: guests # bank
queue: pbatch # partition
reservation: HPCC1B # reservation for this workshop
env:
variables:
OUTPUT_PATH: ./Episode3
study:
- name: amdahl
description: run in parallel
run:
# Here's where we include our MPI wrapper:
cmd: |
$(LAUNCHER) amdahl --terse -p .999 >> amdahl.json
nodes: 1
procs: 4
walltime: "00:00:30"Dry-run (--dry) mode
It’s often useful to run Maestro in --dry mode, which
causes Maestro to create scripts and the directory structure without
actually running jobs. You will see this parameter if you run
maestro run --help.
Challenge
Do a couple dry runs using the script created in the
last challenge. This should help you verify that a new directory
“Episode3” gets created for runs from this episode.
Note: --dry is an input for
maestro run, not for amdahl.
To do a dry run, you shouldn’t need to update your YAML file at all.
Instead, you just run
Key Points
- “Adding
$(LAUNCHER)before commands signals to Maestro to use MPI viasrun.” - “New Maestro runs can be grouped within a new directory specified by
the environment variable
OUTPUT_PATH” - You can use
--dryto verify that the expected directory structure and scripts are created by a given Maestro YAML file.
Content from Placeholders
Last updated on 2024-08-22 | Edit this page
Estimated time: 70 minutes
Overview
Questions
- “How do I make a generic rule?”
Objectives
- “Learn to use variables as placeholders”
- “Learn to run many similar Maestro runs at once”
D.R.Y. (Don’t Repeat Yourself)
Callout
In many programming languages, the bulk of the language features are there to allow the programmer to describe long-winded computational routines as short, expressive, beautiful code. Features in Python, R, or Java, such as user-defined variables and functions are useful in part because they mean we don’t have to write out (or think about) all of the details over and over again. This good habit of writing things out only once is known as the “Don’t Repeat Yourself” principle or D.R.Y.
Maestro YAML files are a form of code and, in any code, repetition can lead to problems (e.g. we rename a data file in one part of the YAML but forget to rename it elsewhere).
In this episode, we’ll set ourselves up with ways to avoid repeating ourselves by using environment variables as placeholders.
Placeholders
Over the course of this lesson, we want to use the
amdahl binary to show how the execution time of a program
changes with the number of processes used. In on our current setup, to
run amdahl for multiple values of procs, we would need to
run our workflow, change procs, rerun, and so forth. We’d
be repeating our workflow a lot, so let’s first try fixing that by
defining multiple rules.
At the end of our last episode, our amdahl.yaml file
contained the sections
YML
(...)
env:
variables:
OUTPUT_PATH: ./Episode3
study:
- name: amdahl
description: run in parallel
run:
# Here's where we include our MPI wrapper:
cmd: |
$(LAUNCHER) amdahl --terse -p .999 >> amdahl.json
nodes: 1
procs: 4
walltime: "00:00:30"Let’s call our existing step amdahl-1 (name
under study) and create a second step called
amdahl-2 which is exactly the same, except that it will
define procs: 8. While we’re at it, let’s update
OUTPUT_PATH so that it is ./Episode4.
The updated part of the script now looks like
YML
(...)
env:
variables:
OUTPUT_PATH: ./Episode4
study:
- name: amdahl-1
description: run in parallel
run:
cmd: |
$(LAUNCHER) amdahl --terse -p .999 >> amdahl.json
nodes: 1
procs: 4
walltime: "00:00:30"
- name: amdahl-2
description: run in parallel
run:
cmd: |
$(LAUNCHER) amdahl --terse -p .999 >> amdahl.json
nodes: 1
procs: 8
walltime: "00:00:30"Challenge
Update amdahl.yaml to include the new info shown above.
Run a dry run to see what your output directory structure looks
like.
Now let’s start to get rid of some of the redundancy in our new workflow.
First off, defining the parallel proportion (-p .999) in
two places makes our lives harder. Now if we want to change this value,
we have to update it in two places, but we can make this easier by using
an environment variable.
Let’s create another environment variable in the
variables second under env. We can define a
new parallel proportion as P: .999. Then, under
run’s cmd for each step, we can call this
environment variable with the syntax $(P).
$(P) holds the place of and will be substituted by
.999 when Maestro creates a Slurm submission script for
us.
Let’s also create an environment variable for our output file,
amdahl.json called OUTPUT and then call that
variable from our cmd fields.
Our updated section will now look like this:
YML
(...)
env:
variables:
P: .999
OUTPUT_PATH: ./Episode4
OUTPUT: amdahl.json
study:
- name: amdahl-1
description: run in parallel
run:
cmd: |
$(LAUNCHER) amdahl --terse -p $(P) >> $(OUTPUT)
nodes: 1
procs: 4
walltime: "00:00:30"
- name: amdahl-2
description: run in parallel
run:
cmd: |
$(LAUNCHER) amdahl --terse -p $(P) >> $(OUTPUT)
nodes: 1
procs: 8
walltime: "00:00:30"We’ve added two new placeholders to make our YAML script to make it a
tad bit more efficient. Note that we had already been using a
placeholder given to us by Maestro: $(LAUNCHER) holds the place of a
call to srun <insert resource requests>.
Callout
Note that in the context of Maestro, the general term for the
“placeholders” declared in the env block are “tokens”.
Challenge
Run your updated amdahl.yaml and check results, to
verify your workflow is working with the changes you’ve made so far.
The full YAML text is
YML
description:
name: Amdahl
description: Run a parallel program
batch:
type: slurm
host: ruby # machine
bank: guests # bank
queue: pbatch # partition
reservation: HPCC1B # reservation for this workshop
env:
variables:
P: .999
OUTPUT_PATH: ./Episode4
OUTPUT: amdahl.json
study:
- name: amdahl-1
description: run in parallel
run:
cmd: |
$(LAUNCHER) amdahl --terse -p $(P) >> $(OUTPUT)
nodes: 1
procs: 4
walltime: "00:00:30"
- name: amdahl-2
description: run in parallel
run:
cmd: |
$(LAUNCHER) amdahl --terse -p $(P) >> $(OUTPUT)
nodes: 1
procs: 8
walltime: "00:00:30"Maestro’s global.parameters
We’re almost ready to perform our scaling study – to see how the
execution time changes as we use more processors in the job.
Unfortunately, we’re still repeating ourselves a lot because, in spite
of the environment variables we created, most of the information defined
for steps amdahl-1 and amdahl-2 is the same.
Only the procs field changes!
A great way to avoid repeating ourselves here by defining a
parameter that lists multiple values of tasks and runs
a separate job step for each value. We do this by adding a
global.parameters section at the bottom of the script. We
then define individual parameters within this section. Each parameter
includes a list of values (Each element is used in its own
job step.) and a label. (The label helps
define how the output directory structure is named.)
This is what it looks like to define a global parameter:
Note that the label should include %% as above; the
%% is itself a placeholder! The directory created for the
output of each job step will be identified by the value of each
parameter it used, and the parameter’s value will be inserted to replace
the %%.
Next, we should update the line under run ->
cmd defining procs to include the name of the
parameter enclosed in $():
If we make this change for steps amdahl-1 and
amdahl-2, they will now look exactly the same, so
we can simply condense them to one step.
The full YAML file will look like
YML
description:
name: Amdahl
description: Run a parallel program
batch:
type: slurm
host: ruby # machine
bank: guests # bank
queue: pbatch # partition
reservation: HPCC1B # reservation for this workshop
env:
variables:
P: .999
OUTPUT: amdahl.json
OUTPUT_PATH: ./Episode4
study:
- name: amdahl
description: run in parallel
run:
# Here's where we include our MPI wrapper:
cmd: |
$(LAUNCHER) amdahl --terse -p $(P) >> $(OUTPUT)
nodes: 1
procs: $(TASKS)
walltime: "00:00:30"
global.parameters:
TASKS:
values: [2, 4, 8, 18, 24, 36]
label: TASKS.%%Challenge
Run maestro run --dry amdahl.yaml using the above YAML
file and investigate the resulting directory structure. How does the
list of task values under global.parameters change the
output directory organization?
Under your current working directory, you should see a directory
structure created with the following format –
Episode4/Amdahl_<Date>-<Time>/amdahl. Within
the amdahl subdirectory, you should see one output
directory for each of the values listed for TASKS under
global.parameters:
OUTPUT
TASKS.18 TASKS.2 TASKS.24 TASKS.36 TASKS.4 TASKS.8Each TASKS... subdirectory will contain the slurm
submission script to be used if this maestro job is run:
OUTPUT
amdahl_TASKS.18.slurm.shRun
before moving on to the next episode, to generate the results for
various task numbers. You’ll be able to see your jobs queuing and
running via squeue -u <username>.
Key Points
- Environment variables are placeholders defined under the
envsection of a Maestro YAML. - Parameters defined under
global.parametersrequire lists of values and labels.
Content from Chaining rules
Last updated on 2024-06-04 | Edit this page
Estimated time: 70 minutes
Overview
Questions
- How do I combine steps into a workflow?
- How do I make a step that uses the outputs from a previous step?
Objectives
- Create workflow pipelines with dependencies.
- Use the outputs of one step as inputs to the next.
A pipeline of multiple rules
In this episode, we will plot the scaling results generated in the
last episode using the script plot_terse_amdahl_results.py.
These results report how long the work of running amdahl
took when using between 2 and 36 processes.
We want to plot these results automatically, i.e. as part of our workflow. In order to do this correctly, we need to make sure that our python plotting script runs only after we have finished calculating all results.
We can control the order and relative timing of when steps execute by defining dependencies.
For example, consider the following YAML,
depends.yaml:
YML
description:
name: Dependency-exploration
description: Experiment with adding dependencies
batch:
type: slurm
host: ruby # machine
bank: guests # bank
queue: pbatch # partition
reservation: HPCC1B # reservation for this workshop
env:
variables:
OUTPUT_PATH: ./Episode5
OUTPUT: date.txt
study:
- name: date-login
description: Write the date and login node's hostname to a file
run:
cmd: |
echo "From login node:" >> $(OUTPUT)
hostname >> $(OUTPUT)
date >> $(OUTPUT)
sleep 10
- name: date-batch
description: Write the date and node's hostname to a file
run:
cmd: |
echo "From batch node:" >> $(OUTPUT)
hostname >> $(OUTPUT)
date >> $(OUTPUT)
sleep 10
nodes: 1
procs: 1
walltime: "00:00:30"This script has two steps, each of which writes the type of node of
its host, the particular hostname, and the date/time before waiting for
10 seconds. The step date-login is run on a login node and
the step date-batch is run on a “batch” or “compute”
node.
Challenge
Run the above script with
You can then cat the output of both steps to
stdout via a command of the form
where {FILL} should be replaced by the date/time info of
this run. This will print the output of the two steps to
stdout.
Which step is printed first and how close together are they completed?
You’ll likely see output somewhat similar to
OUTPUT
From batch node:
pascal16
Tue Mar 26 14:36:04 PDT 2024
From login node:
pascal83
Tue Mar 26 14:36:03 PDT 2024though the times/dates and hostnames will be different. We didn’t try to control the order of operations, so each step will have run as soon as it had resources. Probably you’ll see that the timestamp on the login node is earlier because it will not have to wait for resources from the queue.
Next, let’s add a dependency to ensure that the batch step runs
before the login node step. To add a dependency a line
with the following format must be added to a step’s run
block:
{STEP NAME} is replaced by the name of the step from the
study that you want the current step to depend upon.
If we update date-login to include a dependency, we’ll
see
YML
- name: date-login
description: Write the date and login node's hostname to a file
run:
cmd: |
echo "From login node:" >> $(OUTPUT)
hostname >> $(OUTPUT)
date >> $(OUTPUT)
sleep 10
depends: [date-batch]Now date-login will not run until
date-batch has finished.
Challenge
Update depends.yaml to make date-login wait
for date-batch to complete before running. Then rerun
maestro run depends.yaml.
How has the output of the two date.txt files
changed?
This time, you should see that the date printed from the login node is at least 10 seconds later than the date printed on the batch node. For example, on Pascal I see
OUTPUT
From batch node:
pascal16
Tue Mar 26 15:09:54 PDT 2024
From login node:
pascal83
Tue Mar 26 15:10:53 PDT 2024Callout
Slurm can also be used to define dependencies. How is using Maestro to define the order of our steps any different?
One difference is that we can use Maestro to order steps that
are not seen by Slurm. Above, date-login was run
on the login node. It wasn’t submitted to the queue, and Slurm never saw
that step. (It wasn’t given a Slurm job ID, for example.) Maestro can
control the order of steps running both in and outside the batch queue,
whereas Slurm can only enforce dependencies between Slurm-scheduled
jobs.
A step that waits for all iterations of its dependency
Let’s return to our Amdahl scaling study and the YAML with which we ended in the last episode:
YML
description:
name: Amdahl
description: Run a parallel program
batch:
type: slurm
host: ruby # machine
bank: guests # bank
queue: pbatch # partition
reservation: HPCC1B # reservation for this workshop
env:
variables:
P: .999
OUTPUT: amdahl.json
OUTPUT_PATH: ./Episode5
study:
- name: amdahl
description: run in parallel
run:
# Here's where we include our MPI wrapper:
cmd: |
$(LAUNCHER) amdahl --terse -p $(P) >> $(OUTPUT)
nodes: 1
procs: $(TASKS)
walltime: "00:00:30"
global.parameters:
TASKS:
values: [2, 4, 8, 18, 24, 36]
label: TASKS.%%Ultimately we want to add a plotting step that depends upon
amdahl, but for now let’s create a placeholder that will go
under study and beneath amdahl:
YML
- name: plot
description: Create a plot from `amdahl` results
run:
# We'll update this `cmd` later
cmd: |
echo "This is where we plot"Based on what we saw before, we might think that we just need to add
to the end of this block. Let’s try this to see what happens.
Doing a dry run with amdahl.yaml (text below) should
generate an Amdahl_{Date & time stamp} directory with a
subdirectory for the plot step. Within the ‘plot’
subdirectory, there will be several TASKS.%% subdirectories
– one for each of the values of TASKS defined under
global.parameters in amdahl.yaml. For
example,
OUTPUT
~/Episode5/Amdahl_20240429-153515OUTPUT
amdahl Amdahl.study.pkl batch.info meta plot
Amdahl.pkl Amdahl.txt logs pascal-amdahl.yaml status.csvOUTPUT
TASKS.18 TASKS.2 TASKS.24 TASKS.36 TASKS.4 TASKS.8OUTPUT
TASKS.18/plot_TASKS.18.slurm.shEach of these TASKS.%% subdirectories – such as
plot/TASKS.18 – represents a separate workflow step that is
related to its corresponding amdahl/TASKS.%% workflow step
and directory.
The text of the yaml used is below.
YML
description:
name: Amdahl
description: Run a parallel program
batch:
type: slurm
host: ruby # machine
bank: guests # bank
queue: pbatch # partition
reservation: HPCC1B # reservation for this workshop
env:
variables:
P: .999
OUTPUT: amdahl.json
OUTPUT_PATH: ./Episode5
study:
- name: amdahl
description: run in parallel
run:
# Here's where we include our MPI wrapper:
cmd: |
$(LAUNCHER) amdahl --terse -p $(P) >> $(OUTPUT)
nodes: 1
procs: $(TASKS)
walltime: "00:00:30"
- name: plot
description: Create a plot from `amdahl` results
run:
# We'll update this `cmd` later
cmd: |
echo "This is where we plot"
depends: [amdahl]
global.parameters:
TASKS:
values: [2, 4, 8, 18, 24, 36]
label: TASKS.%%The takeaway from the above challenge is that, a step depending upon
a parameterized step will become parameterized by default. In this case,
creating a plotting step that depends on the amdahl step
will lead to a series of plots, each of which will use data only from a
single run of amdahl.
This is not what we want! Instead, we want to generate a plot that
uses data from several runs of amdahl – each of which will
use a different number of tasks. This means that plot
cannot run until all runs of amdahl have completed.
So, the syntax for defining dependency will change when parameterized
steps are involved. To indicate that we want plot to run
after ALL amdahl steps, we’ll add a
_* to the end of the step name:
Now our new step definition will look like
YML
- name: plot
description: Create a plot from `amdahl` results
run:
# We'll update this `cmd` later
cmd: |
echo "This is where we plot"
depends: [amdahl_*]Challenge
Update your amdahl.yaml so that plot runs
after amdahl has run with all values of TASKS.
Perform a dry run to verify that plot will run only
once.
Your new directory structure should look something like
OUTPUT
amdahl Amdahl.study.pkl batch.info meta plot
Amdahl.pkl Amdahl.txt logs pascal-amdahl.yaml status.csvOUTPUT
plot.slurm.shIn other words, the directory plot will have no
subdirectories.
Using the outputs from a previous step
Manually plotting scaling results
In your working directory, you should have a copy of
plot_terse_amdahl_results.py. The syntax for running this
script is
Before trying to add this command to our workflow, let’s run it
manually to see how it works. We can call the output image
output.jpg. As for the input names, we can use the
.json files created at the end of episode 4. In particular
if you run
using the <Date> and <Time> of
your last run in Episode 4, you should see a list of files named
amdahl.json:
OUTPUT
Episode4/Amdahl_20240326-155434/amdahl/TASKS.18/amdahl.json
Episode4/Amdahl_20240326-155434/amdahl/TASKS.24/amdahl.json
Episode4/Amdahl_20240326-155434/amdahl/TASKS.2/amdahl.json
Episode4/Amdahl_20240326-155434/amdahl/TASKS.36/amdahl.json
Episode4/Amdahl_20240326-155434/amdahl/TASKS.4/amdahl.json
Episode4/Amdahl_20240326-155434/amdahl/TASKS.8/amdahl.jsonYou can use this same filepath with wildcards to specify this list of JSON files as inputs to our python script:
BASH
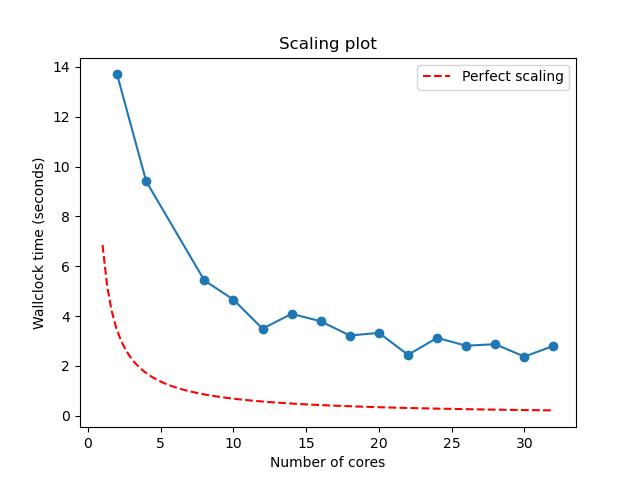
python3 plot_terse_amdahl_results.py output.jpg Episode4/Amdahl_<Date>_<Time>/amdahl/TASKS.*/amdahl.jsonChallenge
Generate a scaling plot by manually specifying the JSON files
produced from a previous run of amdahl.yaml.
The resulting JPEG should look something like
Adding plotting to our workflow
Let’s update our plot step in amdahl.yaml
to include python plotting rather than a placeholder echo
command. We want the updated step to look something like
YML
- name: plot
description: Create a plot from `amdahl` results
run:
cmd: |
python3 plot_terse_amdahl_results.py output.jpg Episode5/Amdahl_<Date>_<Time>/amdahl/TASKS.*/amdahl.json
depends: [amdahl_*]The trouble is that we don’t know the exact value of
Episode5/Amdahl_<Date>_<Time>/amdahl for a job
that we haven’t run yet. Luckily Maestro gives us a placeholder to the
equivalent of this path for the current job –
$(amdahl.workspace). This is the workspace for the
amdahl step, where all outputs for amdahl,
including our TASKS.* directories, are written.
This means we can update the plot step as follows:
YML
- name: plot
description: Create a plot from `amdahl` results
run:
cmd: |
python3 plot_terse_amdahl_results.py output.jpg $(amdahl.workspace)/TASKS.*/amdahl.json
depends: [amdahl_*]Callout
Where does the plot_terse_amdahl_results.py script live?
In Maestro, $(SPECROOT) specifies the root directory from
which you originally ran maestro run .... This is where
plot_terse_amdahl_results.py should live, so let’s be more
precise:
Challenge
Update amdahl.yaml so that
- one step definition runs
amdahlfor 50% parallelizable code using [2, 4, 8, 16, 32] tasks - a second step plots the results.
Your YAML file should look something like
YML
description:
name: Amdahl
description: Run a parallel program
batch:
type: slurm
host: ruby # machine
bank: guests # bank
queue: pbatch # partition
reservation: HPCC1B # reservation for this workshop
env:
variables:
P: .5
OUTPUT: amdahl.json
OUTPUT_PATH: ./Episode5
study:
- name: run-amdahl
description: run in parallel
run:
# Here's where we include our MPI wrapper:
cmd: |
$(LAUNCHER) amdahl --terse -p $(P) >> $(OUTPUT)
nodes: 1
procs: $(TASKS)
walltime: "00:01:30"
- name: plot
description: Create a plot from `amdahl` results
run:
cmd: |
python3 $(SPECROOT)/plot_terse_amdahl_results.py output.jpg $(run-amdahl.workspace)/TASKS.*/amdahl.json
depends: [amdahl_*]
global.parameters:
TASKS:
values: [2, 4, 8, 16, 32]
label: TASKS.%%Errors are normal
Don’t be disheartened if you see errors when first testing your new
Maestro pipelines. There is a lot that can go wrong when writing a new
workflow, and you’ll normally need several iterations to get things just
right. Luckily, Maestro will do some checks for consistency
at the outset of the run. If you specify a dependency that doesn’t exist
(because of a rename, for example), the job will fail before submitting
work to the queue.
Key Points
- We can control the order of steps run in a study by creating dependencies.
- You can create a dependency with the
depends: [{step name}]syntax. - Dependency syntax changes to
depends: [{step name}_*]for parameterized steps.
Content from Multiple parameters
Last updated on 2024-06-04 | Edit this page
Estimated time: 80 minutes
Overview
Questions
- “How do I specify multiple parameters?”
- “How do multiple parameters interact?”
Objectives
- Create scaling results for different proportions of parallelizable code.
- Create and compare scalability plots for codes with different amounts of parallel work.
Adding a second parameter
In this episode, we want to vary P, the fraction of
parallel code, as part of our workflow. To do this, we will add a second
entry under global.parameters and remove the definition for
P under env:
YML
(...)
env:
variables:
OUTPUT: amdahl.json
OUTPUT_PATH: ./Episode6
(...)
global.parameters:
TASKS:
values: [2, 4, 8, 16, 32]
label: TASKS.%%
P:
values: [<Insert values>]
label: P.%%How many values do we want to include for P? We need to
have the same number of values listed for all
global.parameters. This means that to run the previous
scaling study with P=.5, and five values for
TASKS, our global parameters section would specify
.5 for P five times:
YML
global.parameters:
TASKS:
values: [2, 4, 8, 16, 32]
label: TASKS.%%
P:
values: [.5, .5, .5, .5, .5]
label: P.%%Let’s say we want to perform the same scaling study for a second
value of P, .99. This means that we’d have to repeat the
same 5 values for TASKS and then provide the second value
of P 5 times:
YML
global.parameters:
TASKS:
values: [2, 4, 8, 16, 32, 2, 4, 8, 16, 32]
label: TASKS.%%
P:
values: [.5, .5, .5, .5, .5, .99, .99, .99, .99, .99]
label: P.%%At this point, our entire YAML file should look something like
YML
description:
name: Amdahl
description: Run a parallel program
batch:
type: slurm
host: ruby # machine
bank: guests # bank
queue: pbatch # partition
reservation: HPCC1B # reservation for this workshop
env:
variables:
OUTPUT: amdahl.json
OUTPUT_PATH: ./Episode6
study:
- name: run-amdahl
description: run in parallel
run:
# Here's where we include our MPI wrapper:
cmd: |
$(LAUNCHER) amdahl --terse -p $(P) >> $(OUTPUT)
nodes: 1
procs: $(TASKS)
walltime: "00:01:30"
- name: plot
description: Create a plot from `amdahl` results
run:
cmd: |
python3 $(SPECROOT)/plot_terse_amdahl_results.py output.jpg $(run-amdahl.workspace)/TASKS.*/amdahl.json
depends: [amdahl_*]
global.parameters:
TASKS:
values: [2, 4, 8, 16, 32, 2, 4, 8, 16, 32]
label: TASKS.%%
P:
values: [.5, .5, .5, .5, .5, .99, .99, .99, .99, .99]
label: P.%%Challenge
Run the workflow above. Do you generate output.jpg? If
not, why not?
If you use the YAML above, your amdahl-run steps should
work, but your plot step will fail. plot’s
failure will be evident both because output.jpg will be
missing from the plot subdirectory and
because the plot.*.err file in the same directory will
contain an error:
OUTPUT
Traceback (most recent call last):
File "plot_terse_amdahl_results.py", line 46, in <module>
process_files(filenames, output=output)
File "plot_terse_amdahl_results.py", line 10, in process_files
with open(filename, 'r') as file:
FileNotFoundError: [Errno 2] No such file or directory: '~/Episode6/Amdahl_20240328-163359/run-amdahl/TASKS.*/amdahl.json'The problem is that the directory path for .json files
has changed. This will be discussed more below!
The trouble with the YAML above is that our output directory
structure changed when we added a second global parameter, but we didn’t
update the directory path specified under plot.
If we look inside the run-amdahl output folder
(identified as $(run-amdahl.workspace) in our workflow
YAML), its subdirectory names now include information about both global
parameters:
OUTPUT
P.0.5.TASKS.16 P.0.5.TASKS.8 P.0.99.TASKS.4
P.0.5.TASKS.2 P.0.99.TASKS.16 P.0.99.TASKS.8
P.0.5.TASKS.32 P.0.99.TASKS.2
P.0.5.TASKS.4 P.0.99.TASKS.32whereas directory path for our .json files is specified
as $(run-amdahl.workspace)/TASKS.*/amdahl.json under the
plot step.
We could get the plot step to work by simply adding a
wildcard, *, in front of TASKS so that the
path to .json files would be
and the definition for plot would be
YML
- name: plot
description: Create a plot from `amdahl` results
run:
cmd: |
python3 $(SPECROOT)/plot_terse_amdahl_results.py output.jpg $(run-amdahl.workspace)/*TASKS.*/amdahl.json
depends: [amdahl_*]This would allow plot to terminate happily and to
produce an output.jpg file, but that image would plot
output from all .json files as a single line, and we
wouldn’t be able to tell which data points corresponded to a parallel
fraction, \(P=0.85\) and which
corresponded to \(P=0.99\).
If we can generate two plots – one for each value of P –
we’ll more clearly be able to see scaling behavior for these two
situations. We can generate two separate plots by calling
python3 plot_terse_amdahl_results.py ... on two sets of
input files – those in the P.0.5.TASKS* subdirectories of
$(run-amdahl.workspace) and those in the
P.0.99.TASKS* subdirectories.
That means we can generate these two plots by inserting the variable
$(P) into the path –
making the definition for plot
YML
- name: plot
description: Create a plot from `amdahl` results
run:
cmd: |
python3 $(SPECROOT)/plot_terse_amdahl_results.py output.jpg $(run-amdahl.workspace)/P.$(P).TASKS.*/amdahl.json
depends: [amdahl_*]Challenge
Modify your workflow as discussed above to generate output plots for two different values of P. Open these plots and verify they are different from each other. How does changing the workflow to generate two separate plots change the directory structure?
(Feel free to use .5 and .99 or to modify to other values of your choosing.)
Your full YAML file should be similar to
YML
description:
name: Amdahl
description: Run a parallel program
batch:
type: slurm
host: ruby # machine
bank: guests # bank
queue: pbatch # partition
reservation: HPCC1B # reservation for this workshop
env:
variables:
OUTPUT: amdahl.json
OUTPUT_PATH: ./Episode6
study:
- name: run-amdahl
description: run in parallel
run:
# Here's where we include our MPI wrapper:
cmd: |
$(LAUNCHER) amdahl --terse -p $(P) >> $(OUTPUT)
nodes: 1
procs: $(TASKS)
walltime: "00:01:30"
- name: plot
description: Create a plot from `amdahl` results
run:
cmd: |
python3 $(SPECROOT)/plot_terse_amdahl_results.py output.jpg $(run-amdahl.workspace)/P.$(P).TASKS.*/amdahl.json
depends: [amdahl_*]
global.parameters:
TASKS:
values: [2, 4, 8, 16, 32, 2, 4, 8, 16, 32]
label: TASKS.%%
P:
values: [.5, .5, .5, .5, .5, .99, .99, .99, .99, .99]
label: P.%%Modifying plot to include $(P) caused this
step to run twice. As a result, two subdirectories under
plot were created – one for each value of P:
OUTPUT
P.0.5 P.0.99Callout
Instead of modifying the path to our amdahl.json files
to $(run-amdahl.workspace)/P.$(P).TASKS.*/amdahl.json, we
could have equivalently updated it to
$(run-amdahl.workspace)/$(P.label).TASKS.*/amdahl.json.
In other words, P.$(P) is equivalent to
$(P.label). Similarly, in Maestro,
TASKS.$(TASKS) is equivalent to
$(TASKS.label). This syntax works for every global
parameter in Maestro.
Key Points
- “Multiple parameters can be defined under
global.parameters.” - “Lists of values for all global parameters must have the same length; the Nth entries in the lists of values for all global parameters are used in a single job.”